Difference between revisions of "HAEM5:Acute erythroid leukaemia"
| [checked revision] | [checked revision] |
Bailey.Glen (talk | contribs) |
Bailey.Glen (talk | contribs) |
||
| (2 intermediate revisions by the same user not shown) | |||
| Line 4: | Line 4: | ||
{{Under Construction}} | {{Under Construction}} | ||
| − | <blockquote class='blockedit'>{{Box-round|title= | + | <blockquote class='blockedit'>{{Box-round|title=Content Update To WHO 5th Edition Classification Is In Process; Content Below is Based on WHO 4th Edition Classification|This page was converted to the new template on 2023-12-07. The original page can be found at [[HAEM4:Pure Erythroid Leukemia]]. |
Note: need to check if content remains the same | Note: need to check if content remains the same | ||
}}</blockquote> | }}</blockquote> | ||
| − | <span style="color:#0070C0">(General Instructions – The main focus of these pages is the clinically significant genetic alterations in each disease type. Use [https://www.genenames.org/ <u>HUGO-approved gene names and symbols</u>] (italicized when appropriate), [https://varnomen.hgvs.org/ HGVS-based nomenclature for variants], as well as generic names of drugs and testing platforms or assays if applicable. Please complete tables whenever possible and do not delete them (add N/A if not applicable in the table and delete the examples); to add (or move) a row or column to a table, click | + | <span style="color:#0070C0">(General Instructions – The main focus of these pages is the clinically significant genetic alterations in each disease type. Use [https://www.genenames.org/ <u>HUGO-approved gene names and symbols</u>] (italicized when appropriate), [https://varnomen.hgvs.org/ HGVS-based nomenclature for variants], as well as generic names of drugs and testing platforms or assays if applicable. Please complete tables whenever possible and do not delete them (add N/A if not applicable in the table and delete the examples); to add (or move) a row or column to a table, click within the table and select the > symbol that appears to be given options. Please do not delete or alter the section headings. The use of bullet points alongside short blocks of text rather than only large paragraphs is encouraged. Additional instructions below in italicized blue text should not be included in the final page content. Please also see </span><u>[[Author_Instructions]]</u><span style="color:#0070C0"> and [[Frequently Asked Questions (FAQs)|<u>FAQs</u>]] as well as contact your [[Leadership|<u>Associate Editor</u>]] or [mailto:CCGA@cancergenomics.org <u>Technical Support</u>])</span> |
==Primary Author(s)*== | ==Primary Author(s)*== | ||
| Line 17: | Line 17: | ||
__TOC__ | __TOC__ | ||
| − | == | + | ==WHO Classification of Disease== |
| − | + | {| class="wikitable" | |
| − | + | !Structure | |
| − | + | !Disease | |
| − | + | |- | |
| − | + | |Book | |
| + | |Haematolymphoid Tumours (5th ed.) | ||
| + | |- | ||
| + | |Category | ||
| + | |Myeloid proliferations and neoplasms | ||
| + | |- | ||
| + | |Family | ||
| + | |Acute myeloid leukaemia | ||
| + | |- | ||
| + | |Type | ||
| + | |Acute myeloid leukaemia, defined by differentiation | ||
| + | |- | ||
| + | |Subtype(s) | ||
| + | |Acute erythroid leukaemia | ||
| + | |} | ||
==Definition / Description of Disease== | ==Definition / Description of Disease== | ||
| Line 57: | Line 71: | ||
| − | <blockquote class='blockedit'>{{Box-round|title= | + | <blockquote class='blockedit'>{{Box-round|title=v4:Clinical Features|The content below was from the old template. Please incorporate above.}} |
PEL has an aggressive clinical course with neoplastic proliferation of immature erythroid precursor (proerythroblastic or undifferentiated) cells. Average survival rate is three months<ref name=":0" /><ref name=":9" />. PEL is characterized by neoplastic proliferation composed of >80% immature erythroid precursors of which proerythroblasts constitute ≥30%<ref name=":11" />. Clinical features include profound anemia, circulating erythroblasts, pancytopenia, extensive bone marrow involvement, fatigue, infections, weight loss, fever, night sweats, hemoglobin level under 10.0 g/dL, and thrombocytopenia<ref name=":0" /><ref name=":9" />. | PEL has an aggressive clinical course with neoplastic proliferation of immature erythroid precursor (proerythroblastic or undifferentiated) cells. Average survival rate is three months<ref name=":0" /><ref name=":9" />. PEL is characterized by neoplastic proliferation composed of >80% immature erythroid precursors of which proerythroblasts constitute ≥30%<ref name=":11" />. Clinical features include profound anemia, circulating erythroblasts, pancytopenia, extensive bone marrow involvement, fatigue, infections, weight loss, fever, night sweats, hemoglobin level under 10.0 g/dL, and thrombocytopenia<ref name=":0" /><ref name=":9" />. | ||
| Line 111: | Line 125: | ||
| − | <blockquote class='blockedit'>{{Box-round|title= | + | <blockquote class='blockedit'>{{Box-round|title=v4:Chromosomal Rearrangements (Gene Fusions)|The content below was from the old template. Please incorporate above.}} |
The genetic abnormalities that have been identified in PEL are similar to that of AML and MDS and consists of complex chromosomal abnormalities including -5/del(5q), -7/del(7q), +8 and/or RUNX1 and TP53 mutations<ref name=":0" />. Rearrangement of NFIA-CBFA2T3 with t(1;16)(p31;q24) and MYND8-RELA with t(11;20)(p11;q11) have been reported in rare cases<ref name=":9" />. A complex karyotype with 46,XY,der(5)del(5)(p15.1p15.1)t(5;12;7)(p15.1;p13;q32),der(7)t(5;12;7),der(12)del(12)p13p13)t(5;12;7),del(13)(q12q14) was reported in a two year old boy with PEL<ref name=":10" />. | The genetic abnormalities that have been identified in PEL are similar to that of AML and MDS and consists of complex chromosomal abnormalities including -5/del(5q), -7/del(7q), +8 and/or RUNX1 and TP53 mutations<ref name=":0" />. Rearrangement of NFIA-CBFA2T3 with t(1;16)(p31;q24) and MYND8-RELA with t(11;20)(p11;q11) have been reported in rare cases<ref name=":9" />. A complex karyotype with 46,XY,der(5)del(5)(p15.1p15.1)t(5;12;7)(p15.1;p13;q32),der(7)t(5;12;7),der(12)del(12)p13p13)t(5;12;7),del(13)(q12q14) was reported in a two year old boy with PEL<ref name=":10" />. | ||
| Line 137: | Line 151: | ||
| − | <blockquote class='blockedit'>{{Box-round|title= | + | <blockquote class='blockedit'>{{Box-round|title=v4:Clinical Significance (Diagnosis, Prognosis and Therapeutic Implications).|Please incorporate this section into the relevant tables found in: |
* Chromosomal Rearrangements (Gene Fusions) | * Chromosomal Rearrangements (Gene Fusions) | ||
* Individual Region Genomic Gain/Loss/LOH | * Individual Region Genomic Gain/Loss/LOH | ||
| Line 193: | Line 207: | ||
|} | |} | ||
| − | <blockquote class='blockedit'>{{Box-round|title= | + | <blockquote class='blockedit'>{{Box-round|title=v4:Genomic Gain/Loss/LOH|The content below was from the old template. Please incorporate above.}} |
Not Applicable | Not Applicable | ||
| Line 235: | Line 249: | ||
|} | |} | ||
| − | <blockquote class='blockedit'>{{Box-round|title= | + | <blockquote class='blockedit'>{{Box-round|title=v4:Characteristic Chromosomal Aberrations / Patterns|The content below was from the old template. Please incorporate above.}} |
| Line 273: | Line 287: | ||
| − | <blockquote class='blockedit'>{{Box-round|title= | + | <blockquote class='blockedit'>{{Box-round|title=v4:Gene Mutations (SNV/INDEL)|The content below was from the old template. Please incorporate above.}} |
JAK2, FLT3, RAS, NPM1, and CEBPA mutations have been reported to be rare in PEL<ref name=":9" /><ref name=":10" /><ref name=":11" />. Intraclonal heterogeneity and founder mutations of TP53 were reported in 92% (11 out of 12 cases) while co-occurrence of TP53 mutation and deletion due to chromosome 17p abnormalities were detected in 73% of PEL cases<ref>{{Cite journal|last=Montalban-Bravo|first=Guillermo|last2=Benton|first2=Christopher B.|last3=Wang|first3=Sa A.|last4=Ravandi|first4=Farhad|last5=Kadia|first5=Tapan|last6=Cortes|first6=Jorge|last7=Daver|first7=Naval|last8=Takahashi|first8=Koichi|last9=DiNardo|first9=Courtney|date=2017|title=More than 1 TP53 abnormality is a dominant characteristic of pure erythroid leukemia|url=https://www.ncbi.nlm.nih.gov/pubmed/28246192|journal=Blood|volume=129|issue=18|pages=2584–2587|doi=10.1182/blood-2016-11-749903|issn=1528-0020|pmc=5418636|pmid=28246192}}</ref>. | JAK2, FLT3, RAS, NPM1, and CEBPA mutations have been reported to be rare in PEL<ref name=":9" /><ref name=":10" /><ref name=":11" />. Intraclonal heterogeneity and founder mutations of TP53 were reported in 92% (11 out of 12 cases) while co-occurrence of TP53 mutation and deletion due to chromosome 17p abnormalities were detected in 73% of PEL cases<ref>{{Cite journal|last=Montalban-Bravo|first=Guillermo|last2=Benton|first2=Christopher B.|last3=Wang|first3=Sa A.|last4=Ravandi|first4=Farhad|last5=Kadia|first5=Tapan|last6=Cortes|first6=Jorge|last7=Daver|first7=Naval|last8=Takahashi|first8=Koichi|last9=DiNardo|first9=Courtney|date=2017|title=More than 1 TP53 abnormality is a dominant characteristic of pure erythroid leukemia|url=https://www.ncbi.nlm.nih.gov/pubmed/28246192|journal=Blood|volume=129|issue=18|pages=2584–2587|doi=10.1182/blood-2016-11-749903|issn=1528-0020|pmc=5418636|pmid=28246192}}</ref>. | ||
| Line 321: | Line 335: | ||
|} | |} | ||
| − | <blockquote class='blockedit'>{{Box-round|title= | + | <blockquote class='blockedit'>{{Box-round|title=v4:Genes and Main Pathways Involved|The content below was from the old template. Please incorporate above.}} |
The molecular mechanism is not completely understood. | The molecular mechanism is not completely understood. | ||
Latest revision as of 17:20, 6 September 2024
Haematolymphoid Tumours (WHO Classification, 5th ed.)
| This page is under construction |
editContent Update To WHO 5th Edition Classification Is In Process; Content Below is Based on WHO 4th Edition ClassificationThis page was converted to the new template on 2023-12-07. The original page can be found at HAEM4:Pure Erythroid Leukemia.Note: need to check if content remains the same
(General Instructions – The main focus of these pages is the clinically significant genetic alterations in each disease type. Use HUGO-approved gene names and symbols (italicized when appropriate), HGVS-based nomenclature for variants, as well as generic names of drugs and testing platforms or assays if applicable. Please complete tables whenever possible and do not delete them (add N/A if not applicable in the table and delete the examples); to add (or move) a row or column to a table, click within the table and select the > symbol that appears to be given options. Please do not delete or alter the section headings. The use of bullet points alongside short blocks of text rather than only large paragraphs is encouraged. Additional instructions below in italicized blue text should not be included in the final page content. Please also see Author_Instructions and FAQs as well as contact your Associate Editor or Technical Support)
Primary Author(s)*
Ashwini Yenamandra PhD FACMG
WHO Classification of Disease
| Structure | Disease |
|---|---|
| Book | Haematolymphoid Tumours (5th ed.) |
| Category | Myeloid proliferations and neoplasms |
| Family | Acute myeloid leukaemia |
| Type | Acute myeloid leukaemia, defined by differentiation |
| Subtype(s) | Acute erythroid leukaemia |
Definition / Description of Disease
In the 2008 WHO classification, Acute Erythroid leukemia (AEL) was classified into two subtypes: Erythroleukemia (erythroid/myeloid) and Pure Erythroid Leukemia (PEL). However, in the 2016 WHO update, Erythroleukemia was merged into myelodysplastic syndrome, while PEL is now the only type of AEL[1][2][3][4][5][6][7][8][9][10][11][12]. PEL is a distinct entity in the World Health Organization (WHO) classification system within the section of HAEM4:Acute Myeloid Leukemia (AML), Not Otherwise Specified. PEL is a rare form of acute leukemia with an aggressive clinical course and is characterized by an uncontrolled proliferation of immature erythroid precursors (proerythroblastic or undifferentiated)[1][2][3][4][5][6][7][8][9][10][11][12].
Synonyms / Terminology
Also known as AML-M6b and Di Guglielmo syndrome due to the recognition of the work of Di Guglielmo[1][2].
Epidemiology / Prevalence
PEL is extremely rare with a small number of reported cases, accounting for 3-5% of AML cases[1][2][10]. Median survival is usually three months[12].
Clinical Features
Put your text here and fill in the table (Instruction: Can include references in the table. Do not delete table.)
| Signs and Symptoms | EXAMPLE: Asymptomatic (incidental finding on complete blood counts)
EXAMPLE: B-symptoms (weight loss, fever, night sweats) EXAMPLE: Fatigue EXAMPLE: Lymphadenopathy (uncommon) |
| Laboratory Findings | EXAMPLE: Cytopenias
EXAMPLE: Lymphocytosis (low level) |
editv4:Clinical FeaturesThe content below was from the old template. Please incorporate above.PEL has an aggressive clinical course with neoplastic proliferation of immature erythroid precursor (proerythroblastic or undifferentiated) cells. Average survival rate is three months[1][10]. PEL is characterized by neoplastic proliferation composed of >80% immature erythroid precursors of which proerythroblasts constitute ≥30%[12]. Clinical features include profound anemia, circulating erythroblasts, pancytopenia, extensive bone marrow involvement, fatigue, infections, weight loss, fever, night sweats, hemoglobin level under 10.0 g/dL, and thrombocytopenia[1][10].
Sites of Involvement
Bone marrow, Blood
Morphologic Features
PEL is characterized by medium to large erythroblasts with round nuclei, fine chromatin and one or more nucleoli (proerythroblast). Cytoplasm is deeply basophilic, often granular with demarcated vacuoles and are often Periodic-Acid-Schiff stain (PAS) positive. Blasts can be small and may resemble lymphoblasts[1]. Cells are usually negative for Myeloperoxidase (MPO) and Sudan Black (SBB). Bone marrow biopsy may have undifferentiated cells[1].
Immunophenotype
Differentiated PEL may express Glycophorin and hemoglobin A, absence of myeloperoxidase (MPO) and other myeloid markers[1]. Blasts are negative for HLA-Dr and CD34 but positive for CD117[1]. Immature forms can be negative for Glycophorin or weekly expressed. Positive for Carbonic anhydrase 1, Gero antobody against the Gerbich blood group or CD36 especially at earlier stages of differentiation. CD41 and CD61 are negative[1][12].
| Finding | Marker |
|---|---|
| Positive (universal) | Hemoglobin A, Glycophorin A, Spectrin, ABH blood group antigens, and HLA-DR |
| Positive (subset) | CD13, CD33, CD34, CD117 (KIT), and MPO, Gerbich blood group (Gero) antibody, carbonic anhydrase 1, and CD36, CD41 and CD61 |
| Negative (universal) | Myeloid-associated markers such as MPO,CD13,CD33,CD61, B and T Cell markers -CD10, CD19, CD79a, CD2, CD3, CD5, monocytic markers CD11c CD14
Megakaryoblastic markers: CD61, Others: CD34, anti-kappa, anti-lambda, CD45 |
| Negative (subset) | HLA-DR, CD34, Glycophorin A |
Chromosomal Rearrangements (Gene Fusions)
Put your text here and fill in the table
| Chromosomal Rearrangement | Genes in Fusion (5’ or 3’ Segments) | Pathogenic Derivative | Prevalence | Diagnostic Significance (Yes, No or Unknown) | Prognostic Significance (Yes, No or Unknown) | Therapeutic Significance (Yes, No or Unknown) | Notes |
|---|---|---|---|---|---|---|---|
| EXAMPLE: t(9;22)(q34;q11.2) | EXAMPLE: 3'ABL1 / 5'BCR | EXAMPLE: der(22) | EXAMPLE: 20% (COSMIC)
EXAMPLE: 30% (add reference) |
Yes | No | Yes | EXAMPLE:
The t(9;22) is diagnostic of CML in the appropriate morphology and clinical context (add reference). This fusion is responsive to targeted therapy such as Imatinib (Gleevec) (add reference). |
editv4:Chromosomal Rearrangements (Gene Fusions)The content below was from the old template. Please incorporate above.The genetic abnormalities that have been identified in PEL are similar to that of AML and MDS and consists of complex chromosomal abnormalities including -5/del(5q), -7/del(7q), +8 and/or RUNX1 and TP53 mutations[1]. Rearrangement of NFIA-CBFA2T3 with t(1;16)(p31;q24) and MYND8-RELA with t(11;20)(p11;q11) have been reported in rare cases[10]. A complex karyotype with 46,XY,der(5)del(5)(p15.1p15.1)t(5;12;7)(p15.1;p13;q32),der(7)t(5;12;7),der(12)del(12)p13p13)t(5;12;7),del(13)(q12q14) was reported in a two year old boy with PEL[11].
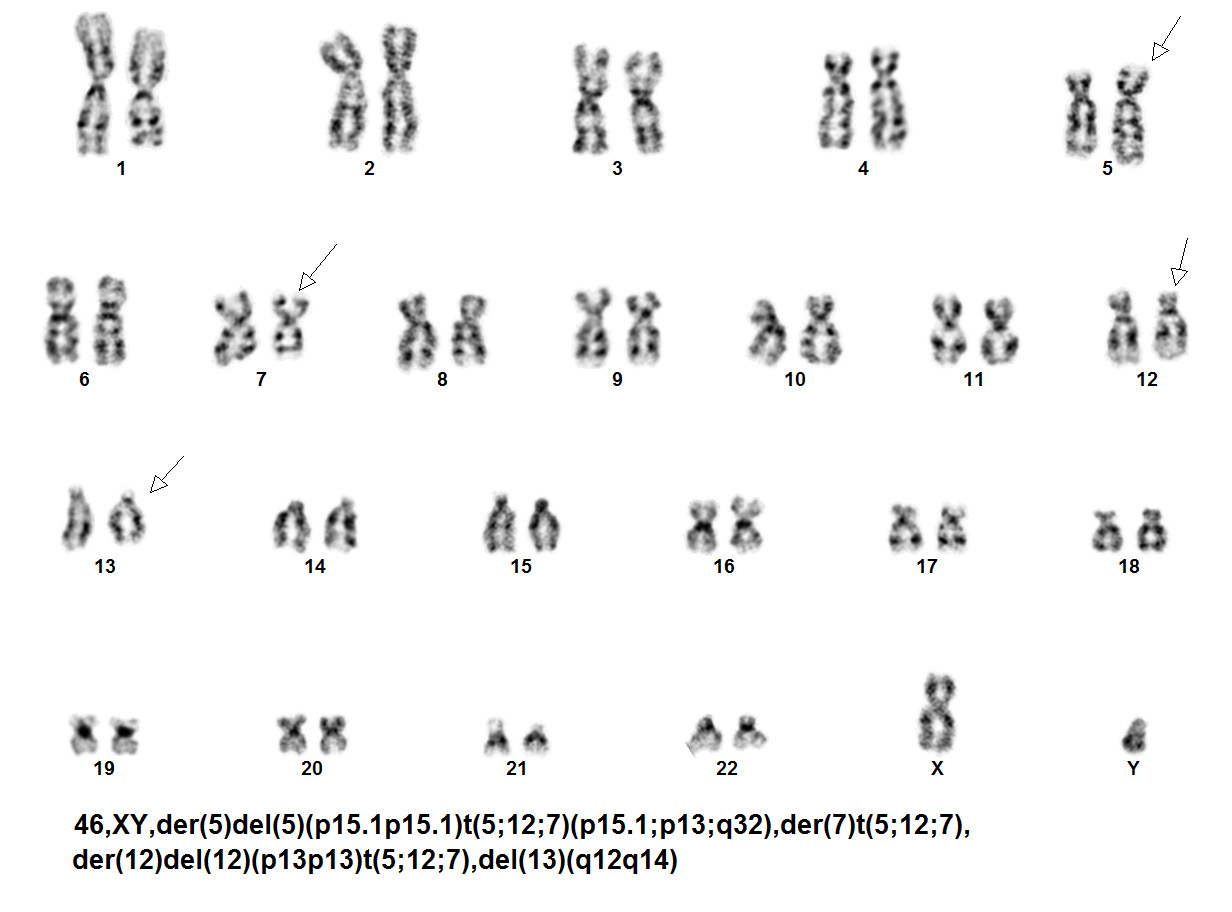 Karyotype of two year old boy with PEL[11].
Karyotype of two year old boy with PEL[11].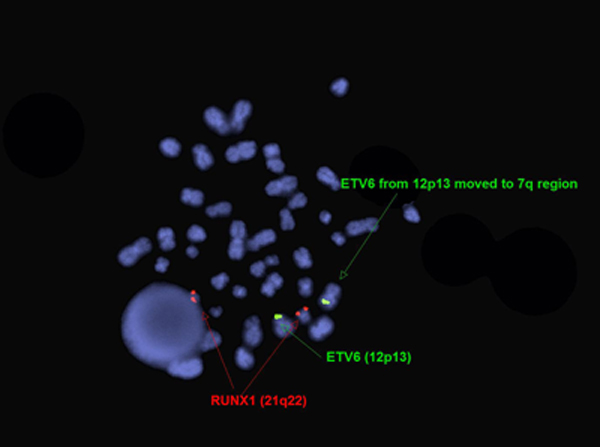 FISH for ETV6 and RUNX1 in two year old boy with PEL[11].
FISH for ETV6 and RUNX1 in two year old boy with PEL[11]. FISH for EGR1 and D5S23,D5S721 (5p-) in two year old boy with PEL[11].
FISH for EGR1 and D5S23,D5S721 (5p-) in two year old boy with PEL[11].
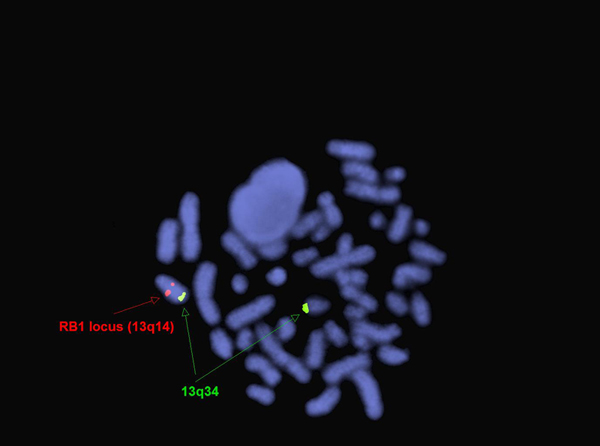 FISH for RB1 and 13q34 (13q deletion) in two year old boy with PEL[11].
FISH for RB1 and 13q34 (13q deletion) in two year old boy with PEL[11].
Chromosomal Rearrangement Genes in Fusion (5’ or 3’ Segments) Pathogenic Derivative Prevalence t(1;16)(p31;q24) 5’NFIA/ 3’CBFA2T3 der(16) Rare t(11;20)(p11;q11) 5’MYND8/ 3’RELA der(11) Rare


.jpg)
.jpg)
editv4:Clinical Significance (Diagnosis, Prognosis and Therapeutic Implications).Please incorporate this section into the relevant tables found in:
- Chromosomal Rearrangements (Gene Fusions)
- Individual Region Genomic Gain/Loss/LOH
- Characteristic Chromosomal Patterns
- Gene Mutations (SNV/INDEL)
PEL has rapid and aggressive clinical course. Patients with PEL are treated similar to other types of AML. Stem cell transplantation (SCT) may have an improvement in the outcome of the disease. No therapeutic agents for specific target pathways are currently available[3].
Individual Region Genomic Gain / Loss / LOH
Put your text here and fill in the table (Instructions: Includes aberrations not involving gene fusions. Can include references in the table. Can refer to CGC workgroup tables as linked on the homepage if applicable. Do not delete table.)
| Chr # | Gain / Loss / Amp / LOH | Minimal Region Genomic Coordinates [Genome Build] | Minimal Region Cytoband | Diagnostic Significance (Yes, No or Unknown) | Prognostic Significance (Yes, No or Unknown) | Therapeutic Significance (Yes, No or Unknown) | Notes |
|---|---|---|---|---|---|---|---|
| EXAMPLE:
7 |
EXAMPLE: Loss | EXAMPLE:
chr7:1- 159,335,973 [hg38] |
EXAMPLE:
chr7 |
Yes | Yes | No | EXAMPLE:
Presence of monosomy 7 (or 7q deletion) is sufficient for a diagnosis of AML with MDS-related changes when there is ≥20% blasts and no prior therapy (add reference). Monosomy 7/7q deletion is associated with a poor prognosis in AML (add reference). |
| EXAMPLE:
8 |
EXAMPLE: Gain | EXAMPLE:
chr8:1-145,138,636 [hg38] |
EXAMPLE:
chr8 |
No | No | No | EXAMPLE:
Common recurrent secondary finding for t(8;21) (add reference). |
editv4:Genomic Gain/Loss/LOHThe content below was from the old template. Please incorporate above.Not Applicable
Chromosome Number Gain/Loss/Amp/LOH Region 5 Loss Whole chromosome or q-arm 7 Loss Whole chromosome or q-arm 8 Gain Whole chromosome 17 Loss P-arm
Characteristic Chromosomal Patterns
Put your text here (EXAMPLE PATTERNS: hyperdiploid; gain of odd number chromosomes including typically chromosome 1, 3, 5, 7, 11, and 17; co-deletion of 1p and 19q; complex karyotypes without characteristic genetic findings; chromothripsis. Do not delete table.)
| Chromosomal Pattern | Diagnostic Significance (Yes, No or Unknown) | Prognostic Significance (Yes, No or Unknown) | Therapeutic Significance (Yes, No or Unknown) | Notes |
|---|---|---|---|---|
| EXAMPLE:
Co-deletion of 1p and 18q |
Yes | No | No | EXAMPLE:
See chromosomal rearrangements table as this pattern is due to an unbalanced derivative translocation associated with oligodendroglioma (add reference). |
editv4:Characteristic Chromosomal Aberrations / PatternsThe content below was from the old template. Please incorporate above.
Gene Mutations (SNV / INDEL)
Put your text here and fill in the table (Instructions: This table is not meant to be an exhaustive list; please include only genes/alterations that are recurrent and common as well as either disease defining and/or clinically significant. Can include references in the table. For clinical significance, denote associations with FDA-approved therapy (not an extensive list of applicable drugs) and NCCN or other national guidelines if applicable. Can also refer to CGC workgroup tables as linked on the homepage if applicable as well as any high impact papers or reviews of gene mutations in this entity. Do not delete table.)
| Gene; Genetic Alteration | Presumed Mechanism (Tumor Suppressor Gene [TSG] / Oncogene / Other) | Prevalence (COSMIC / TCGA / Other) | Concomitant Mutations | Mutually Exclusive Mutations | Diagnostic Significance (Yes, No or Unknown) | Prognostic Significance (Yes, No or Unknown) | Therapeutic Significance (Yes, No or Unknown) | Notes |
|---|---|---|---|---|---|---|---|---|
| EXAMPLE: TP53; Variable LOF mutations
EXAMPLE: EGFR; Exon 20 mutations EXAMPLE: BRAF; Activating mutations |
EXAMPLE: TSG | EXAMPLE: 20% (COSMIC)
EXAMPLE: 30% (add Reference) |
EXAMPLE: IDH1 R123H | EXAMPLE: EGFR amplification | EXAMPLE: Excludes hairy cell leukemia (HCL) (add reference).
|
Note: A more extensive list of mutations can be found in cBioportal (https://www.cbioportal.org/), COSMIC (https://cancer.sanger.ac.uk/cosmic), ICGC (https://dcc.icgc.org/) and/or other databases. When applicable, gene-specific pages within the CCGA site directly link to pertinent external content.
editv4:Gene Mutations (SNV/INDEL)The content below was from the old template. Please incorporate above.JAK2, FLT3, RAS, NPM1, and CEBPA mutations have been reported to be rare in PEL[10][11][12]. Intraclonal heterogeneity and founder mutations of TP53 were reported in 92% (11 out of 12 cases) while co-occurrence of TP53 mutation and deletion due to chromosome 17p abnormalities were detected in 73% of PEL cases[13].
Gene Mutation Oncogene/Tumor Suppressor/Other Presumed Mechanism (LOF/GOF/Other; Driver/Passenger) Prevalence (COSMIC/TCGA/Other) EXAMPLE: TP53 EXAMPLE: R273H EXAMPLE: Tumor Suppressor EXAMPLE: LOF EXAMPLE: 20% Other Mutations
Type Gene/Region/Other Concomitant Mutations EXAMPLE: IDH1 R123H Secondary Mutations EXAMPLE: Trisomy 7 Mutually Exclusive EXAMPLE: EGFR Amplification
Epigenomic Alterations
Not Applicable
Genes and Main Pathways Involved
Put your text here and fill in the table (Instructions: Can include references in the table. Do not delete table.)
| Gene; Genetic Alteration | Pathway | Pathophysiologic Outcome |
|---|---|---|
| EXAMPLE: BRAF and MAP2K1; Activating mutations | EXAMPLE: MAPK signaling | EXAMPLE: Increased cell growth and proliferation |
| EXAMPLE: CDKN2A; Inactivating mutations | EXAMPLE: Cell cycle regulation | EXAMPLE: Unregulated cell division |
| EXAMPLE: KMT2C and ARID1A; Inactivating mutations | EXAMPLE: Histone modification, chromatin remodeling | EXAMPLE: Abnormal gene expression program |
editv4:Genes and Main Pathways InvolvedThe content below was from the old template. Please incorporate above.The molecular mechanism is not completely understood.
Genetic Diagnostic Testing Methods
Morphology and IHC.
Familial Forms
Not Applicable
Additional Information
Differential Diagnosis: PEL without morphological differentiation of erythroid maturation can be difficult to distinguish from megakaryoblastic leukemia (AML), ALL or lymphoma. The erythroid precursor immunophenotype helps in the diagnosis. Some cases can be complex with concurrent erythroid megakaryocytic involvement[1].
Links
References
(use the "Cite" icon at the top of the page) (Instructions: Add each reference into the text above by clicking on where you want to insert the reference, selecting the “Cite” icon at the top of the page, and using the “Automatic” tab option to search such as by PMID to select the reference to insert. The reference list in this section will be automatically generated and sorted. If a PMID is not available, such as for a book, please use the “Cite” icon, select “Manual” and then “Basic Form”, and include the entire reference.)
- ↑ 1.00 1.01 1.02 1.03 1.04 1.05 1.06 1.07 1.08 1.09 1.10 1.11 1.12 Arber DA, et al., (2008). World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th edition. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, Editors. IARC Press: Lyon, France, p135-136.
- ↑ 2.0 2.1 2.2 2.3 Qiu, Shaowei; et al. (2017). "An analysis of 97 previously diagnosed de novo adult acute erythroid leukemia patients following the 2016 revision to World Health Organization classification". BMC cancer. 17 (1): 534. doi:10.1186/s12885-017-3528-6. ISSN 1471-2407. PMC 5550989. PMID 28793875.
- ↑ 3.0 3.1 3.2 Zuo, Zhuang; et al. (2010). "Acute erythroid leukemia". Archives of Pathology & Laboratory Medicine. 134 (9): 1261–1270. doi:10.1043/2009-0350-RA.1. ISSN 1543-2165. PMID 20807044.
- ↑ 4.0 4.1 Arber, Daniel A.; et al. (2016). "The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia". Blood. 127 (20): 2391–2405. doi:10.1182/blood-2016-03-643544. ISSN 1528-0020. PMID 27069254.
- ↑ 5.0 5.1 Zuo, Zhuang; et al. (2012). "Acute myeloid leukemia (AML) with erythroid predominance exhibits clinical and molecular characteristics that differ from other types of AML". PloS One. 7 (7): e41485. doi:10.1371/journal.pone.0041485. ISSN 1932-6203. PMC 3402404. PMID 22844482.
- ↑ 6.0 6.1 Grossmann, V.; et al. (2013). "Acute erythroid leukemia (AEL) can be separated into distinct prognostic subsets based on cytogenetic and molecular genetic characteristics". Leukemia. 27 (9): 1940–1943. doi:10.1038/leu.2013.144. ISSN 1476-5551. PMID 23648669.
- ↑ 7.0 7.1 Porwit, Anna; et al. (2011). "Acute myeloid leukemia with expanded erythropoiesis". Haematologica. 96 (9): 1241–1243. doi:10.3324/haematol.2011.050526. ISSN 1592-8721. PMC 3166090. PMID 21880638.
- ↑ 8.0 8.1 Hasserjian, Robert P.; et al. (2010). "Acute erythroid leukemia: a reassessment using criteria refined in the 2008 WHO classification". Blood. 115 (10): 1985–1992. doi:10.1182/blood-2009-09-243964. ISSN 1528-0020. PMC 2942006. PMID 20040759.
- ↑ 9.0 9.1 Wang, Sa A.; et al. (2015). "Acute Erythroleukemias, Acute Megakaryoblastic Leukemias, and Reactive Mimics: A Guide to a Number of Perplexing Entities". American Journal of Clinical Pathology. 144 (1): 44–60. doi:10.1309/AJCPRKYAT6EZQHC7. ISSN 1943-7722. PMID 26071461.
- ↑ 10.0 10.1 10.2 10.3 10.4 10.5 10.6 Wang, Wei; et al. (2017). "Pure erythroid leukemia". American Journal of Hematology. 92 (3): 292–296. doi:10.1002/ajh.24626. ISSN 1096-8652. PMID 28006859.
- ↑ 11.0 11.1 11.2 11.3 11.4 11.5 11.6 11.7 A, Yenamandra (2016). "Acute Erythroblastic Leukemia (AEL): A Rare Subset of De Novo AML with A Complex Rearrangement Involving ETV6 Locus and Loss of RB1 Locus". International Clinical Pathology Journal. 2 (2). doi:10.15406/icpjl.2016.02.00032.
- ↑ 12.0 12.1 12.2 12.3 12.4 12.5 Fs, Khan (2017). "Pure Erythroid Leukemia: The Sole Acute Erythroid Leukemia". International Journal of Bone Marrow Research. 1 (1): 001–005. doi:10.29328/journal.ijbmr.1001001.
- ↑ Montalban-Bravo, Guillermo; et al. (2017). "More than 1 TP53 abnormality is a dominant characteristic of pure erythroid leukemia". Blood. 129 (18): 2584–2587. doi:10.1182/blood-2016-11-749903. ISSN 1528-0020. PMC 5418636. PMID 28246192.
Notes
*Primary authors will typically be those that initially create and complete the content of a page. If a subsequent user modifies the content and feels the effort put forth is of high enough significance to warrant listing in the authorship section, please contact the CCGA coordinators (contact information provided on the homepage). Additional global feedback or concerns are also welcome. *Citation of this Page: “Acute erythroid leukaemia”. Compendium of Cancer Genome Aberrations (CCGA), Cancer Genomics Consortium (CGC), updated 09/6/2024, https://ccga.io/index.php/HAEM5:Acute_erythroid_leukaemia.