Difference between revisions of "HAEM5:Myelodysplastic neoplasm with biallelic TP53 inactivation"
| [unchecked revision] | [checked revision] |
Bailey.Glen (talk | contribs) |
Bailey.Glen (talk | contribs) |
||
| (2 intermediate revisions by the same user not shown) | |||
| Line 1: | Line 1: | ||
{{DISPLAYTITLE:Myelodysplastic neoplasm with biallelic TP53 inactivation}} | {{DISPLAYTITLE:Myelodysplastic neoplasm with biallelic TP53 inactivation}} | ||
| − | [[HAEM5:Table_of_Contents|Haematolymphoid Tumours (5th ed.)]] | + | [[HAEM5:Table_of_Contents|Haematolymphoid Tumours (WHO Classification, 5th ed.)]] |
| − | |||
| − | |||
| − | |||
| − | |||
==Primary Author(s)*== | ==Primary Author(s)*== | ||
| + | Eric McGinnis, MD, Vancouver General Hospital | ||
| + | ==WHO Classification of Disease== | ||
| − | + | {| class="wikitable" | |
| − | + | !Structure | |
| − | + | !Disease | |
| − | + | |- | |
| − | + | |Book | |
| − | + | |Haematolymphoid Tumours (5th ed.) | |
| − | + | |- | |
| − | + | |Category | |
| − | + | |Myeloid proliferations and neoplasms | |
| − | + | |- | |
| − | + | |Family | |
| + | |Myelodysplastic neoplasms | ||
| + | |- | ||
| + | |Type | ||
| + | |Myelodysplastic neoplasms, with defining genetic abnormalities | ||
| + | |- | ||
| + | |Subtype(s) | ||
| + | |Myelodysplastic neoplasm with biallelic TP53 inactivation | ||
| + | |} | ||
==Definition / Description of Disease== | ==Definition / Description of Disease== | ||
| + | Myelodysplastic neoplasm with biallelic ''TP53'' inactivation (MDS-bi''TP53'') is a myeloid neoplasm which is characterized by ineffective hematopoiesis, manifesting clinically and pathologically as cytopenia(s), bone marrow morphologic dysplasia, and propensity to progress to acute myeloid leukemia, and which carries either two ''TP53'' mutations or a single mutation accompanied by either evidence of concurrent ''TP53'' deletion or copy-neutral loss of heterozygosity (LOH); in the appropriate setting, a pathogenic ''TP53'' variant with high variant allele fraction (VAF > 49%) can be considered presumptive evidence of biallelic inactivation status.<ref>{{Cite journal|last=Khoury|first=Joseph D.|last2=Solary|first2=Eric|last3=Abla|first3=Oussama|last4=Akkari|first4=Yassmine|last5=Alaggio|first5=Rita|last6=Apperley|first6=Jane F.|last7=Bejar|first7=Rafael|last8=Berti|first8=Emilio|last9=Busque|first9=Lambert|date=2022-07|title=The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms|url=https://pubmed.ncbi.nlm.nih.gov/35732831|journal=Leukemia|volume=36|issue=7|pages=1703–1719|doi=10.1038/s41375-022-01613-1|issn=1476-5551|pmc=9252913|pmid=35732831}}</ref><ref name=":2" /> | ||
| − | + | MDS bi-''TP53'' supersedes other genetically and morphologically defined MDS entities. By definition, in addition to the requirement for evidence of biallelic ''TP53'' inactivation, general criteria for diagnosis of MDS must be met and diagnostic criteria for acute myeloid leukemia must not be met. Acute erythroid leukemia, in which ''TP53'' mutations are common, must be specifically excluded by morphologic and/or immunophenotypic assessment.<ref name=":2" /> | |
| − | |||
==Synonyms / Terminology== | ==Synonyms / Terminology== | ||
| − | + | Myelodysplastic neoplasm with multi-hit ''TP53'' inactivation, MDS-bi''TP53'' | |
| − | |||
| − | |||
==Epidemiology / Prevalence== | ==Epidemiology / Prevalence== | ||
| + | Inactivating mutations or losses of ''TP53'', whether monoallelic or biallelic, have been reported to occur in approximately 11% of MDS, with a higher frequency in therapy-related MDS (18%) relative to ''de novo'' MDS (6%).<ref name=":0">{{Cite journal|last=Bernard|first=Elsa|last2=Nannya|first2=Yasuhito|last3=Hasserjian|first3=Robert P.|last4=Devlin|first4=Sean M.|last5=Tuechler|first5=Heinz|last6=Medina-Martinez|first6=Juan S.|last7=Yoshizato|first7=Tetsuichi|last8=Shiozawa|first8=Yusuke|last9=Saiki|first9=Ryunosuke|date=2020-10|title=Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes|url=https://pubmed.ncbi.nlm.nih.gov/32747829|journal=Nature Medicine|volume=26|issue=10|pages=1549–1556|doi=10.1038/s41591-020-1008-z|issn=1546-170X|pmc=8381722|pmid=32747829}}</ref> These genomic aberrations resulting in loss of ''TP53'' are present in a biallelic or multi-hit state in two-thirds of patients with MDS (with a higher reported frequency of 84% in patients with therapy-related disease).<ref name=":0" /> The incidence of MDS-bi''TP53'' has been estimated to be approximately 0.03 persons per 100,000 per year.<ref name=":2">Germing U., Claudi M., Zerbini N., et al. Myelodysplastic neoplasm with biallelic TP53 inactivation. In: WHO Classification of Tumours Editorial Board. Haematolymphoid tumours [Internet; beta version ahead of print]. Lyon (France): International Agency for Research on Cancer; 2022 [cited YYYY Mmm D]. (WHO classification of tumours series, 5th ed.; vol. 11). Available from: <nowiki>https://tumourclassification.iarc.who.int/chapters/63</nowiki>.</ref> | ||
| + | ==Clinical Features== | ||
| + | Signs and symptoms are typically nonspecific and related to cytopenias (unified thresholds for cytopenias in WHO5 diagnostic categories are detailed below, though these must be interpreted in the context of laboratory reference ranges, patient sex, and relevant comorbidities). ''TP53'' mutations show a strong association with thrombocytopenia in MDS.<ref name=":0" /><ref name=":1">{{Cite journal|last=Haase|first=Detlef|last2=Stevenson|first2=Kristen E.|last3=Neuberg|first3=Donna|last4=Maciejewski|first4=Jaroslaw P.|last5=Nazha|first5=Aziz|last6=Sekeres|first6=Mikkael A.|last7=Ebert|first7=Benjamin L.|last8=Garcia-Manero|first8=Guillermo|last9=Haferlach|first9=Claudia|date=2019-07|title=TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups|url=https://pubmed.ncbi.nlm.nih.gov/30635634|journal=Leukemia|volume=33|issue=7|pages=1747–1758|doi=10.1038/s41375-018-0351-2|issn=1476-5551|pmc=6609480|pmid=30635634}}</ref> | ||
| − | + | *Anemia (hemoglobin < 130 g/L in males, < 120 g/L in females) | |
| − | + | **Pallor, fatigue | |
| − | + | *Neutropenia (absolute neutrophil count < 1.8x10<sup>9</sup>/L) | |
| + | **Infection | ||
| + | *Thrombocytopenia (platelet count < 150x10<sup>9</sup>/L) | ||
| + | **Bruising, petechiae, bleeding | ||
| − | + | MDS-bi''TP53'' is a high-risk disease, with increased risk of leukemic transformation and decreased overall survival; this elevated risk is independent of IPSS-R risk stratification and persists in the context of allogeneic hematopoietic stem cell transplantation. In the absence of comprehensive assessment for copy number losses or copy neutral LOH affecting the ''TP53'' locus, evidence suggests that the presence of a ''TP53'' mutation at high (>40%) VAF, particularly in the context of a complex karyotype, may be associated with a similarly poor prognosis to MDS-bi''TP53''.<ref name=":0" /><ref name=":2" /><ref>{{Cite journal|last=Sallman|first=D. A.|last2=Komrokji|first2=R.|last3=Vaupel|first3=C.|last4=Cluzeau|first4=T.|last5=Geyer|first5=S. M.|last6=McGraw|first6=K. L.|last7=Al Ali|first7=N. H.|last8=Lancet|first8=J.|last9=McGinniss|first9=M. J.|date=2016-03|title=Impact of TP53 mutation variant allele frequency on phenotype and outcomes in myelodysplastic syndromes|url=https://pubmed.ncbi.nlm.nih.gov/26514544|journal=Leukemia|volume=30|issue=3|pages=666–673|doi=10.1038/leu.2015.304|issn=1476-5551|pmc=7864381|pmid=26514544}}</ref><ref>{{Cite journal|last=Grob|first=Tim|last2=Al Hinai|first2=Adil S. A.|last3=Sanders|first3=Mathijs A.|last4=Kavelaars|first4=François G.|last5=Rijken|first5=Melissa|last6=Gradowska|first6=Patrycja L.|last7=Biemond|first7=Bart J.|last8=Breems|first8=Dimitri A.|last9=Maertens|first9=Johan|date=2022-04-14|title=Molecular characterization of mutant TP53 acute myeloid leukemia and high-risk myelodysplastic syndrome|url=https://pubmed.ncbi.nlm.nih.gov/35108372|journal=Blood|volume=139|issue=15|pages=2347–2354|doi=10.1182/blood.2021014472|issn=1528-0020|pmid=35108372}}</ref> | |
{| class="wikitable" | {| class="wikitable" | ||
|'''Signs and Symptoms''' | |'''Signs and Symptoms''' | ||
| − | | | + | |Pallor |
| − | + | Fatigue | |
| − | + | Infection | |
| − | + | Bruising | |
| + | |||
| + | Bleeding | ||
| + | |||
| + | Petechiae | ||
|- | |- | ||
|'''Laboratory Findings''' | |'''Laboratory Findings''' | ||
| − | | | + | |Anemia (hemoglobin < 130 g/L in males, < 120 g/L in females) |
| − | + | Neutropenia (neutrophils < 1.8x10<sup>9</sup>/L) | |
| + | |||
| + | Thrombocytopenia (platelets < 150x10<sup>9</sup>/L) | ||
| + | |||
| + | Circulating blasts (+/-) | ||
| + | |||
| + | Elevated lactate dehydrogenase (+/-) | ||
|} | |} | ||
==Sites of Involvement== | ==Sites of Involvement== | ||
| + | Bone marrow and peripheral blood; rarely, extramedullary proliferations occur.<ref>{{Cite journal|last=Fan|first=N.|last2=Lavu|first2=S.|last3=Hanson|first3=C. A.|last4=Tefferi|first4=A.|date=2018-11-19|title=Extramedullary hematopoiesis in the absence of myeloproliferative neoplasm: Mayo Clinic case series of 309 patients|url=https://pubmed.ncbi.nlm.nih.gov/30455416|journal=Blood Cancer Journal|volume=8|issue=12|pages=119|doi=10.1038/s41408-018-0156-6|issn=2044-5385|pmc=6242913|pmid=30455416}}</ref> | ||
| + | ==Morphologic Features== | ||
| + | Morphologic dysplasia is the hallmark of MDS, including MDS-bi''TP53''; by definition, at least one lineage . ''TP53'' mutations in MDS have been associated with particularly severe granulocytic dysplasia and with increased frequency of bone marrow blasts.<ref name=":0" /><ref name=":1" /><ref>{{Cite journal|last=Della Porta|first=M. G.|last2=Travaglino|first2=E.|last3=Boveri|first3=E.|last4=Ponzoni|first4=M.|last5=Malcovati|first5=L.|last6=Papaemmanuil|first6=E.|last7=Rigolin|first7=G. M.|last8=Pascutto|first8=C.|last9=Croci|first9=G.|date=2015-01|title=Minimal morphological criteria for defining bone marrow dysplasia: a basis for clinical implementation of WHO classification of myelodysplastic syndromes|url=https://pubmed.ncbi.nlm.nih.gov/24935723|journal=Leukemia|volume=29|issue=1|pages=66–75|doi=10.1038/leu.2014.161|issn=1476-5551|pmid=24935723}}</ref> ''TP53'' mutations are enriched in MDS associated with bone marrow fibrosis.<ref>{{Cite journal|last=Duarte|first=Fernando B.|last2=Barbosa|first2=Maritza C.|last3=Jesus Dos Santos|first3=Talyta E.|last4=Lemes|first4=Romélia P. G.|last5=Vasconcelos|first5=João P.|last6=de Vasconcelos|first6=Paulo R. L.|last7=Rocha|first7=Francisco D.|last8=Zalcberg|first8=Ilana|last9=Coutinho|first9=Diego F.|date=2018-05|title=Bone marrow fibrosis at diagnosis is associated with TP53 overexpression and adverse prognosis in low-risk myelodysplastic syndrome|url=https://pubmed.ncbi.nlm.nih.gov/28318026|journal=British Journal of Haematology|volume=181|issue=4|pages=547–549|doi=10.1111/bjh.14656|issn=1365-2141|pmid=28318026}}</ref><ref>{{Cite journal|last=Loghavi|first=Sanam|last2=Al-Ibraheemi|first2=Alyaa|last3=Zuo|first3=Zhuang|last4=Garcia-Manero|first4=Guillermo|last5=Yabe|first5=Mariko|last6=Wang|first6=Sa A.|last7=Kantarjian|first7=Hagop M.|last8=Yin|first8=Cameron C.|last9=Miranda|first9=Roberto N.|date=2015-10|title=TP53 overexpression is an independent adverse prognostic factor in de novo myelodysplastic syndromes with fibrosis|url=https://pubmed.ncbi.nlm.nih.gov/26123119|journal=British Journal of Haematology|volume=171|issue=1|pages=91–99|doi=10.1111/bjh.13529|issn=1365-2141|pmc=5577911|pmid=26123119}}</ref> | ||
| + | {| class="wikitable" | ||
| + | !Hematopoietic lineage | ||
| + | !Characteristic dysplastic features | ||
| + | |- | ||
| + | |'''Erythroid''' | ||
| + | |Nuclear budding | ||
| + | Internuclear bridging | ||
| − | + | Multinuclearity | |
| − | + | Karyorrhexis | |
| − | + | Megaloblastosis | |
| − | + | Cytoplasmic vacuolation | |
| − | + | Ring sideroblasts | |
| − | + | Periodic acid Schiff positivity | |
|- | |- | ||
| − | + | |'''Granulocytic''' | |
| + | |Nuclear hyposegmentation (Pelgeroid changes) | ||
| + | Nuclear hypersegmentation | ||
| + | |||
| + | Cytoplasmic hypogranularity | ||
| + | |||
| + | Abnormally small cell size | ||
| + | |||
| + | Auer rods | ||
| + | |||
| + | Pseudo-Chédiak–Higashi granules | ||
|- | |- | ||
| − | |Positive (universal)| | + | |'''Megakaryocytic''' |
| + | |Nuclear hypolobation | ||
| + | Widely separated nuclear lobes | ||
| + | |||
| + | Micromegakaryocytes | ||
| + | |} | ||
| + | [[File:Dysplastic_blood_and_bone_marrow_features_in_MDS.tif|alt=|thumb|1300x1300px|Multilineage dysplasia in MDS. Left: ''TP53'' mutation is associated with severe dysgranulopoiesis, including hypogranular and hypolobated neutrophils (black arrows). Center: Dysgranulopoiesis (arrows) and dyserythropoiesis with basophilic stippling and nuclear atypia (arrowheads). Right: Dysmegakaryopoiesis with separated nuclear lobes and small hypolobated cells (white arrows). Image from: Eric McGinnis, Vancouver General Hospital|none]] | ||
| + | <br />[[File:Bone marrow morphology in MDS biTP53.tif|thumb|1300x1300px|MDS-bi''TP53'' is associated with high-grade features, including increased blasts and bone marrow fibrosis, at an increased frequency. Left: Bone marrow showing disorganized hematopoiesis and increased numbers of blasts forming small clusters (white arrows). Center: Immunohistochemical staining for CD34 highlighting blasts in increased number and small dense blast clusters (arrowheads). Right: Bone marrow reticulin fibrosis (black arrows). Image from: Eric McGinnis, Vancouver General Hospital|alt=|none]] | ||
| + | <br /> | ||
| + | ==Immunophenotype== | ||
| + | Immunophenotyping by flow cytometry using assays designed to detect abnormal numbers, light scatter characteristics, and antigen expression patterns of hematopoietic cells can provide useful data supportive of a diagnosis of MDS (and aid in blast enumeration); however, these findings are not specific to MDS-bi''TP53'' and are not formalized in current diagnostic criteria.<ref name=":2" /><ref>{{Cite journal|last=Kern|first=Wolfgang|last2=Westers|first2=Theresia M.|last3=Bellos|first3=Frauke|last4=Bene|first4=Marie Christine|last5=Bettelheim|first5=Peter|last6=Brodersen|first6=Lisa Eidenschink|last7=Burbury|first7=Kate|last8=Chu|first8=Sung-Chao|last9=Cullen|first9=Matthew|date=2023-01|title=Multicenter prospective evaluation of diagnostic potential of flow cytometric aberrancies in myelodysplastic syndromes by the ELN iMDS flow working group|url=https://pubmed.ncbi.nlm.nih.gov/36416672|journal=Cytometry. Part B, Clinical Cytometry|volume=104|issue=1|pages=51–65|doi=10.1002/cyto.b.22105|issn=1552-4957|pmid=36416672}}</ref> | ||
| + | |||
| + | Immunohistochemical evaluation of TP53 protein expression in bone marrow specimens can be used to inform ''TP53'' mutation status. Several studies evaluating TP53 protein expression in bone marrow specimens have consistently demonstrated nuclear overexpression (thought to result from stabilization of TP53 resulting from dominant negative mutations), when present in an increased fraction of cells, to be associated with pathogenic ''TP53'' mutation and to be similarly predictive of poor outcomes.<ref name=":3">{{Cite journal|last=Tashakori|first=Mehrnoosh|last2=Kadia|first2=Tapan|last3=Loghavi|first3=Sanam|last4=Daver|first4=Naval|last5=Kanagal-Shamanna|first5=Rashmi|last6=Pierce|first6=Sherry|last7=Sui|first7=Dawen|last8=Wei|first8=Peng|last9=Khodakarami|first9=Farnoosh|date=2022-07-07|title=TP53 copy number and protein expression inform mutation status across risk categories in acute myeloid leukemia|url=https://pubmed.ncbi.nlm.nih.gov/35390143|journal=Blood|volume=140|issue=1|pages=58–72|doi=10.1182/blood.2021013983|issn=1528-0020|pmc=9346958|pmid=35390143}}</ref><ref>{{Cite journal|last=Saft|first=Leonie|last2=Karimi|first2=Mohsen|last3=Ghaderi|first3=Mehran|last4=Matolcsy|first4=András|last5=Mufti|first5=Ghulam J.|last6=Kulasekararaj|first6=Austin|last7=Göhring|first7=Gudrun|last8=Giagounidis|first8=Aristoteles|last9=Selleslag|first9=Dominik|date=2014-06|title=p53 protein expression independently predicts outcome in patients with lower-risk myelodysplastic syndromes with del(5q)|url=https://pubmed.ncbi.nlm.nih.gov/24682512|journal=Haematologica|volume=99|issue=6|pages=1041–1049|doi=10.3324/haematol.2013.098103|issn=1592-8721|pmc=4040908|pmid=24682512}}</ref><ref>{{Cite journal|last=Loghavi|first=Sanam|last2=Al-Ibraheemi|first2=Alyaa|last3=Zuo|first3=Zhuang|last4=Garcia-Manero|first4=Guillermo|last5=Yabe|first5=Mariko|last6=Wang|first6=Sa A.|last7=Kantarjian|first7=Hagop M.|last8=Yin|first8=Cameron C.|last9=Miranda|first9=Roberto N.|date=2015-10|title=TP53 overexpression is an independent adverse prognostic factor in de novo myelodysplastic syndromes with fibrosis|url=https://pubmed.ncbi.nlm.nih.gov/26123119|journal=British Journal of Haematology|volume=171|issue=1|pages=91–99|doi=10.1111/bjh.13529|issn=1365-2141|pmc=5577911|pmid=26123119}}</ref><ref>{{Cite journal|last=Ruzinova|first=Marianna B.|last2=Lee|first2=Yi-Shan|last3=Duncavage|first3=Eric J.|last4=Welch|first4=John S.|date=2019-08|title=TP53 immunohistochemistry correlates with TP53 mutation status and clearance in decitabine-treated patients with myeloid malignancies|url=https://pubmed.ncbi.nlm.nih.gov/30792212|journal=Haematologica|volume=104|issue=8|pages=e345–e348|doi=10.3324/haematol.2018.205302|issn=1592-8721|pmc=6669137|pmid=30792212}}</ref><ref>{{Cite journal|last=Kubbutat|first=M. H.|last2=Jones|first2=S. N.|last3=Vousden|first3=K. H.|date=1997-05-15|title=Regulation of p53 stability by Mdm2|url=https://pubmed.ncbi.nlm.nih.gov/9153396|journal=Nature|volume=387|issue=6630|pages=299–303|doi=10.1038/387299a0|issn=0028-0836|pmid=9153396}}</ref> Complete absence of TP53 expression demonstrable by immunohistochemistry has similarly been associated with ''TP53'' mutation (typically frameshift, nonsense, and splice site, often with coexisting copy loss).<ref name=":3" /> | ||
| + | <br /> | ||
| + | [[File:TP53 IHC panel.tif|thumb|1300x1300px|Immunohistochemical evaluation of TP53 expression correlates with ''TP53'' mutation status in MDS-bi''TP53''. Left: Normal TP53 expression in MDS without ''TP53'' mutation; cells show variable nuclear staining. Center: TP53 overexpression in MDS with biallelic ''TP53'' inactivation resulting from a missense mutation and loss of 17p; the majority of cells show excessive nuclear staining. Right: Absence of TP53 expression in MDS with biallelic ''TP53'' inactivation resulting from a truncating mutation and loss of 17p; the great majority of cells show absence of any detectable TP53 staining.|alt=|none]] | ||
| + | {| class="wikitable" | ||
| + | |'''Positive (universal)''' | ||
| + | |N/A | ||
|- | |- | ||
| − | |Positive (subset)| | + | |'''Positive (subset)''' |
| + | |N/A | ||
|- | |- | ||
| − | |Negative (universal)| | + | |'''Negative (universal)''' |
| + | |N/A | ||
|- | |- | ||
| − | |Negative (subset)| | + | |'''Negative (subset)''' |
| + | |N/A | ||
|} | |} | ||
==Chromosomal Rearrangements (Gene Fusions)== | ==Chromosomal Rearrangements (Gene Fusions)== | ||
| + | There are no known recurrent rearrangements resulting in gene fusions associated with MDS-bi''TP53''. | ||
| + | {| class="wikitable" | ||
| + | |'''Chromosomal Rearrangement''' | ||
| + | |'''Genes in Fusion''' | ||
| − | + | '''(5’ or 3’ Segments)''' | |
| + | |'''Pathogenic Derivative''' | ||
| + | |'''Prevalence''' | ||
| + | |'''Diagnostic Significance (Yes, No or Unknown)''' | ||
| + | |'''Prognostic Significance (Yes, No or Unknown)''' | ||
| + | |'''Therapeutic Significance (Yes, No or Unknown)''' | ||
| + | |'''Notes''' | ||
| + | |- | ||
| + | |<span class="blue-text">EXAMPLE:</span> t(9;22)(q34;q11.2) | ||
| + | |<span class="blue-text">EXAMPLE:</span> 3'ABL1 / 5'BCR | ||
| + | |<span class="blue-text">EXAMPLE:</span> der(22) | ||
| + | |<span class="blue-text">EXAMPLE:</span> 20% (COSMIC) | ||
| − | + | <span class="blue-text">EXAMPLE:</span> 30% (add reference) | |
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
|Yes | |Yes | ||
|No | |No | ||
|Yes | |Yes | ||
| − | |EXAMPLE | + | |<span class="blue-text">EXAMPLE:</span> |
| − | The t(9;22) is diagnostic of CML in the appropriate morphology and clinical context (add reference). This fusion is responsive to targeted therapy such as Imatinib (Gleevec) (add reference). | + | The t(9;22) is diagnostic of CML in the appropriate morphology and clinical context (add reference). This fusion is responsive to targeted therapy such as Imatinib (Gleevec) (add reference). |
| − | |} | + | |} |
| − | |||
| − | |||
| − | + | ==Individual Region Genomic Gain/Loss/LOH== | |
| + | MDS bi-''TP53'' is strongly associated with karyotype complexity, with a higher average number of cytogenetic abnormalities (rearrangements, copy gains, and copy losses) relative to MDS with unmutated ''TP53'' or with monoallelic mutation, with particular predilection for recurrent copy number losses.<ref name=":1" /><ref name=":0" /> Recurrent chromosomal gains, losses, and regions of LOH described which have been observed at higher frequency in MDS-bi''TP53'' include<ref name=":0" /><ref name=":2" /><ref name=":1" />: | ||
| − | + | '''Loss / Deletion''' | |
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | 7 | + | *Deletion 5q (most common co-occurring abnormality) |
| − | + | *Loss of chromosome 7 or deletion 7q (second most common co-occurring abnormality) | |
| − | + | *Deletion 17p | |
| + | *Deletion 12p | ||
| + | *Losses involving chromosome 13 | ||
| + | *Losses involving chromosome 18 | ||
| + | *Deletion 20q | ||
| − | + | '''Gain''' | |
| − | |||
| − | + | *Trisomy 8 | |
| − | + | *Trisomy 21 | |
| − | + | *Gain of 1p | |
| − | + | *Trisomy 22 | |
| − | + | *Gain of 11q | |
| − | |||
| − | |||
| − | |||
| − | + | Copy-neutral LOH involving chromosome 17p and the ''TP53'' locus is a component of the diagnostic definition of MDS-bi''TP53'' and has been reported to occur in 21% of patients with this disease, usually in the setting of a single ''TP53'' mutation (i.e. not typically in association with more than one distinct ''TP53'' mutation).<ref name=":0" /> It is not currently known whether other instances of recurrent copy-neutral LOH in myeloid neoplasms are observed at significantly higher frequency in MDS-bi''TP53''. | |
| − | | | + | {| class="wikitable" |
| − | | | + | |'''Chr #''' |
| + | |'''Gain/Loss/Amp/LOH''' | ||
| + | |'''Minimal Region Genomic Coordinates [Genome Build]''' | ||
| + | |'''Minimal Region Cytoband''' | ||
| + | |'''Diagnostic Significance (Yes, No or Unknown)''' | ||
| + | |'''Prognostic Significance''' | ||
| − | + | '''(Yes, No or Unknown)''' | |
| − | | | + | |'''Therapeutic Significance''' |
| − | chr8 | + | '''(Yes, No or Unknown)''' |
| + | |'''Notes''' | ||
| + | |- | ||
| + | |1 | ||
| + | |Gain | ||
| + | | | ||
| + | |1p | ||
| + | |No | ||
| + | |No* | ||
| + | |No | ||
| + | | | ||
| + | |- | ||
| + | |5 | ||
| + | |Loss | ||
| + | | | ||
| + | |5q | ||
| + | |No | ||
| + | |No* | ||
| + | |No | ||
| + | |Most common co-occurring abnormality | ||
| + | |- | ||
| + | |7 | ||
| + | |Loss | ||
| + | | | ||
| + | |chr7 / 7q | ||
| + | |No | ||
| + | |No* | ||
| + | |No | ||
| + | |Second most common co-occurring abnormality | ||
| + | |- | ||
| + | |8 | ||
| + | |Gain | ||
| + | | | ||
| + | |chr8 | ||
| + | |No | ||
| + | |No* | ||
| + | |No | ||
| + | | | ||
| + | |- | ||
| + | |11 | ||
| + | |Gain | ||
| + | | | ||
| + | |11q | ||
|No | |No | ||
| + | |No* | ||
|No | |No | ||
| + | | | ||
| + | |- | ||
| + | |12 | ||
| + | |Loss | ||
| + | | | ||
| + | |12p | ||
|No | |No | ||
| − | | | + | |No* |
| − | + | |No | |
| − | + | | | |
| − | | | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
|- | |- | ||
| − | + | |13 | |
| − | + | |Loss | |
| − | + | | | |
| − | + | |13q | |
| − | + | |No | |
| + | |No* | ||
| + | |No | ||
| + | | | ||
|- | |- | ||
| − | | | + | |17 |
| − | + | |Loss / LOH | |
| − | + | | | |
| + | |17p | ||
| + | |Yes | ||
| + | |Yes** | ||
|Yes | |Yes | ||
| + | |Key component of entity classification | ||
| + | |- | ||
| + | |18 | ||
| + | |Loss | ||
| + | | | ||
| + | | | ||
| + | |No | ||
| + | |No* | ||
| + | |No | ||
| + | | | ||
| + | |- | ||
| + | |20 | ||
| + | |Loss | ||
| + | | | ||
| + | |20q | ||
|No | |No | ||
| + | |No* | ||
|No | |No | ||
| − | | | + | | |
| + | |- | ||
| + | |21 | ||
| + | |Gain | ||
| + | | | ||
| + | |chr21 | ||
| + | |No | ||
| + | |No* | ||
| + | |No | ||
| + | | | ||
| + | |- | ||
| + | |22 | ||
| + | |Gain | ||
| + | | | ||
| + | |chr22 | ||
| + | |No | ||
| + | |No* | ||
| + | |No | ||
| + | | | ||
| + | |} | ||
| + | <nowiki>*</nowiki>Adverse prognosis associated with MDS-bi''TP53'' appears to persist independent of IPSS-R status | ||
| − | + | <nowiki>**</nowiki>MDS-bi''TP53'' diagnosis and therefore prognostic stratification is dependent on loss or LOH involving 17p unless multiple mutations are present | |
| − | | | + | |
| − | = | + | ==Characteristic Chromosomal Patterns== |
| + | MDS-bi''TP53'' is strongly associated with complex (and monosomal) karyotypes; patients with biallelic inactivation of ''TP53'' showed significantly higher numbers of distinct cytogenetic abnormalities (median six excluding chromosome 17) relative to patients with monoallelic inactivation (median one), and a large majority of these tend to be accounted for by abnormalities associated with genomic loss (deletion/monosomy), with a median of four such abnormalities in MDS-bi''TP53'', which are particularly enriched for deletions involving chromosome 5q (identified in 85% of patients).<ref name=":0" /><ref name=":2" /><ref name=":1" /> Abnormalities involving chromosome 17p resulting in ''TP53'' loss are, by virtue of diagnostic criteria, present in approximately one third of MDS bi-''TP53'', in which they may result from simple deletion or unbalanced rearrangement, and which may in cases be submicroscopic/cryptic to analysis of banded chromosomes.<ref name=":0" /> | ||
| + | [[File:Complex karyotype with loss of 17p in MDS with biTP53.tif|thumb|1300x1300px|MDS-bi''TP53'' is strongly associated with complex and monosomal karyotypes. Left: Cytogenetic analysis of bone marrow cells in MDS showing a complex karyotype with deletion of 17p and characteristic recurrent abnormalities, including del(5q), del(7q), del(20q), and loss of chromosome 18. Center: Interphase FISH showing deletion of one ''TP53'' locus resulting from the del(17p). Right: Loss of 17p material resulting in deletion of ''TP53'' often results from unbalanced rearrangements involving 17p (top) or deletion of 17p (bottom) in the context of a complex karyotype. | ||
| + | |||
| + | Image from: Eric McGinnis, Vancouver General Hospital|alt=|none]] | ||
| + | {| class="wikitable" | ||
| + | |'''Chromosomal Pattern''' | ||
| + | |'''Diagnostic Significance (Yes, No or Unknown)''' | ||
| + | |'''Prognostic Significance''' | ||
| − | + | '''(Yes, No or Unknown)''' | |
| + | |'''Therapeutic Significance''' | ||
| − | + | '''(Yes, No or Unknown)''' | |
| + | |'''Notes''' | ||
|- | |- | ||
| − | + | |Complex karyotype | |
| − | + | |No (see note) | |
| − | + | |Yes (see note) | |
| − | + | |No (see note) | |
| − | + | |Often with abnormalities resulting in chromosomal losses, especially involving chromosome 5q and chromosome 7 | |
| − | + | Complex karyotypes are strongly associated with MDS-bi''TP53'' and, in the absence of available data for identification of LOH at the ''TP53'' locus, may be suggestive of biallelic inactivation and may be associated with similarly poor prognosis | |
| − | | | + | |} |
| − | + | ==Gene Mutations (SNV/INDEL)== | |
| + | At least one mutation involving ''[[TP53]]'' is required to establish a diagnosis of MDS bi''TP53''; a second distinct mutation involving ''TP53'', fulfilling diagnostic criteria without deletion or copy neutral LOH, is identified in approximately one third of patients with MDS-bi''TP53''.<ref name=":2" /><ref name=":0" /> ''TP53'' mutations reported in MDS-bi''TP53'' show a similar pattern of distribution to those previously reported, primarily clustering in the DNA-binding domain with smaller numbers of (primarily truncating) mutations upstream (extending to the transactivation domain) or downstream (extending downstream of the oligomerization domain); available data suggest approximately 70% of mutations to be missense variants, 28% (a higher frequency than was observed in MDS with monoallelic ''TP53'' mutation) to be truncating (including nonsense, frameshift, splice site, and nonstop variants), and a small fraction to be in-frame deletions or insertions.<ref name=":0" /> In MDS-bi''TP53'', 20% of ''TP53'' variants identified were accounted for by hotspot mutations affecting amino acids 273, 220, 248, and 175 (in order of descending frequency). Variants identified in the context of multiple distinct ''TP53'' mutations have been reported with a median VAF of 32% (range: 2% to 52%) while those associated with a second hit resulting from ''TP53'' locus deletion or copy-neutral LOH have a reported median VAF of 41% (2% to 92% with a bimodal distribution) and 71% (2% to 98%), respectively.<ref name=":0" /> | ||
| − | + | ''TP53''-mutated MDS, in general, tends to carry fewer cooperating somatic mutations that MDS without ''TP53'' mutation; MDS bi-''TP53'' is further associated with fewer cooperating somatic mutations than MDS with monoallelic inactivation of ''TP53'', with 40% of MDS-bi''TP53'' reported to show no additional identifiable driver mutations.<ref name=":0" /><ref name=":1" /> Genes recurrently mutated in this group are similar to those described in MDS in general, and include ''DNMT3A'', ''TET2'', ''ASXL1'', ''U2AF1'', ''PPM1D'', ''EZH2'', ''SF3B1'', ''NF1''; infrequently, comutations identified include ''BCOR'', ''JAK2'', and ''CBL''. MDS carrying at least one mutation in ''TP53'', as a whole, has been observed to less frequently carry mutations in ''ASXL1'', ''U2AF1'', and ''RUNX1'', while MDS-bi''TP53'' has been observed to have lower rates of ''TET2'', ''SF3B1'', ''ASXL1'', ''SRSF2'', and ''RUNX1'' mutation relative to MDS with a single ''TP53'' mutation.<ref name=":0" /><ref name=":1" /> | |
| − | + | Note: A more extensive list of mutations can be found in cBioportal (https://www.cbioportal.org/), COSMIC (https://cancer.sanger.ac.uk/cosmic), ICGC (https://dcc.icgc.org/) and/or other databases. When applicable, gene-specific pages within the CCGA site directly link to pertinent external content. | |
| − | | | + | {| class="wikitable" |
| − | | | + | |'''Gene; Genetic Alteration''' |
| + | |'''Presumed Mechanism (Tumor Suppressor Gene (TSG)/Oncogene/Other)''' | ||
| + | |'''Prevalence (COSMIC/ TCGA/Other)''' | ||
| + | |'''Concomitant Mutations''' | ||
| + | |'''Mutually Exclusive Mutations''' | ||
| + | |'''Diagnostic Significance (Yes, No or Unknown)''' | ||
| + | |'''Prognostic Significance''' | ||
| − | + | '''(Yes, No or Unknown)''' | |
| − | | | + | |'''Therapeutic Significance''' |
| − | | | + | |
| − | | | + | '''(Yes, No or Unknown)''' |
| − | | | + | |'''Notes''' |
| − | | | + | |- |
| − | | | + | |''TP53;'' LOF and/or GOF mutations |
| − | + | |Tumor Suppressor Gene / Oncogene (GOF variants) | |
| + | |100% (entity-defining for MDS-bi''TP53'') | ||
| + | |''DNMT3A, TET2, ASXL1, U2AF1, PPM1D, EZH2, SF3B1, NF1'' (see note) | ||
| + | |None | ||
| + | |Yes | ||
| + | |Yes | ||
| + | |Yes (see note) | ||
| + | |MDS-bi''TP53'' carries, on average, less cooperating somatic mutations than MDS without ''TP53'' mutation | ||
| + | MDS-bi''TP53'' is associated with poorer responses to most therapeutic modalities | ||
|} | |} | ||
| − | |||
==Epigenomic Alterations== | ==Epigenomic Alterations== | ||
| − | + | Similar to other MDS entities, mutations in chromatin-modifying/epigenetic regulator genes (e.g. ''TET2'', ''EZH2'', ''DNMT3A'', ''ASXL1'') are recurrent in MDS-bi''TP53'', albeit at a lower frequency. Certain ''TP53'' mutations, including hotspot mutations identified in MDS-bi''TP53'', have been demonstrated to confer gain-of-function effects to the mutant TP53, some of which appear to be mediated through modification of the activities of chromatin regulatory/epigenetic modifier genes such as ''KMT2A'', ''KMT2D'', and ''EZH2''.<ref>{{Cite journal|last=Zhu|first=Jiajun|last2=Sammons|first2=Morgan A.|last3=Donahue|first3=Greg|last4=Dou|first4=Zhixun|last5=Vedadi|first5=Masoud|last6=Getlik|first6=Matthäus|last7=Barsyte-Lovejoy|first7=Dalia|last8=Al-awar|first8=Rima|last9=Katona|first9=Bryson W.|date=2015-09-10|title=Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth|url=https://pubmed.ncbi.nlm.nih.gov/26331536|journal=Nature|volume=525|issue=7568|pages=206–211|doi=10.1038/nature15251|issn=1476-4687|pmc=4568559|pmid=26331536}}</ref><ref>{{Cite journal|last=Chen|first=Sisi|last2=Wang|first2=Qiang|last3=Yu|first3=Hao|last4=Capitano|first4=Maegan L.|last5=Vemula|first5=Sasidhar|last6=Nabinger|first6=Sarah C.|last7=Gao|first7=Rui|last8=Yao|first8=Chonghua|last9=Kobayashi|first9=Michihiro|date=2019-12-11|title=Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway|url=https://pubmed.ncbi.nlm.nih.gov/31827082|journal=Nature Communications|volume=10|issue=1|pages=5649|doi=10.1038/s41467-019-13542-2|issn=2041-1723|pmc=6906427|pmid=31827082}}</ref> | |
| − | |||
| − | |||
==Genes and Main Pathways Involved== | ==Genes and Main Pathways Involved== | ||
| − | + | p53, a tumour repressor transcription factor encoded by the ''TP53'' gene, is central to regulation of a complex network of pathways by which cells respond to intra- or extracellular stressors, including cell cycle progression and response to hyperproliferation signals, cell survival and apoptosis, and maintenance of genome stability.<ref>{{Cite journal|last=Eischen|first=Christine M.|date=2016-06-01|title=Genome Stability Requires p53|url=https://pubmed.ncbi.nlm.nih.gov/27252396|journal=Cold Spring Harbor Perspectives in Medicine|volume=6|issue=6|pages=a026096|doi=10.1101/cshperspect.a026096|issn=2157-1422|pmc=4888814|pmid=27252396}}</ref><ref>{{Cite journal|last=Levine|first=Arnold J.|last2=Oren|first2=Moshe|date=2009-10|title=The first 30 years of p53: growing ever more complex|url=https://pubmed.ncbi.nlm.nih.gov/19776744|journal=Nature Reviews. Cancer|volume=9|issue=10|pages=749–758|doi=10.1038/nrc2723|issn=1474-1768|pmc=2771725|pmid=19776744}}</ref><ref>{{Cite journal|last=Boettcher|first=Steffen|last2=Miller|first2=Peter G.|last3=Sharma|first3=Rohan|last4=McConkey|first4=Marie|last5=Leventhal|first5=Matthew|last6=Krivtsov|first6=Andrei V.|last7=Giacomelli|first7=Andrew O.|last8=Wong|first8=Waihay|last9=Kim|first9=Jesi|date=2019-08-09|title=A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies|url=https://pubmed.ncbi.nlm.nih.gov/31395785|journal=Science (New York, N.Y.)|volume=365|issue=6453|pages=599–604|doi=10.1126/science.aax3649|issn=1095-9203|pmc=7327437|pmid=31395785}}</ref> Mutations inactivating ''TP53'' typically result in disruption of normal tumour suppressor activities of the protein and, depending on the cell state and mutational profile, may exert dominant negative effects on residual wild-type p53 and/or may confer neomorphic oncogenic function to the protein.<ref>{{Cite journal|last=Stein|first=Yan|last2=Aloni-Grinstein|first2=Ronit|last3=Rotter|first3=Varda|date=2020-12-31|title=Mutant p53 oncogenicity: dominant-negative or gain-of-function?|url=https://pubmed.ncbi.nlm.nih.gov/33159515|journal=Carcinogenesis|volume=41|issue=12|pages=1635–1647|doi=10.1093/carcin/bgaa117|issn=1460-2180|pmid=33159515}}</ref><ref>{{Cite journal|last=Boettcher|first=Steffen|last2=Miller|first2=Peter G.|last3=Sharma|first3=Rohan|last4=McConkey|first4=Marie|last5=Leventhal|first5=Matthew|last6=Krivtsov|first6=Andrei V.|last7=Giacomelli|first7=Andrew O.|last8=Wong|first8=Waihay|last9=Kim|first9=Jesi|date=2019-08-09|title=A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies|url=https://pubmed.ncbi.nlm.nih.gov/31395785|journal=Science (New York, N.Y.)|volume=365|issue=6453|pages=599–604|doi=10.1126/science.aax3649|issn=1095-9203|pmc=7327437|pmid=31395785}}</ref> p53 is required for genomic stability and mutations affecting ''TP53'' are among the most frequent abnormalities identified in human malignancies, in which they are strongly associated with genomic instability (in the form of increased frequency of aneuploidy, polyploidy/tetraploidy, and chromosomal rearrangements).<ref name=":0" /><ref>{{Cite journal|last=Eischen|first=Christine M.|date=2016-06-01|title=Genome Stability Requires p53|url=https://pubmed.ncbi.nlm.nih.gov/27252396|journal=Cold Spring Harbor Perspectives in Medicine|volume=6|issue=6|pages=a026096|doi=10.1101/cshperspect.a026096|issn=2157-1422|pmc=4888814|pmid=27252396}}</ref> | |
| − | + | {| class="wikitable" | |
| − | {| class="wikitable | + | |'''Gene; Genetic Alteration''' |
| − | | | + | |'''Pathway''' |
| − | + | |'''Pathophysiologic Outcome''' | |
|- | |- | ||
| − | | | + | |''TP53''; Inactivating +/- Gain of abnormal function mutations |
| − | | | + | |TP53 signaling network; cell cycle arrest, DNA repair, cell senescence, apoptosis in response to DNA damage, oncogene activation, cell stressors |
| − | + | |Failures of DNA repair, cell cycle arrest, apoptosis in response to DNA damage, hyperproliferation, or cell stressors | |
| − | |||
| − | |||
| − | |||
| − | | | ||
| − | |||
| − | |||
| − | |||
| − | |||
|} | |} | ||
| + | |||
==Genetic Diagnostic Testing Methods== | ==Genetic Diagnostic Testing Methods== | ||
| + | As classification of MDS-bi''TP53'' requires demonstration of at least one ''TP53'' mutation, sequencing analysis is required in all cases (i.e. cytogenetic evidence supportive of ''TP53'' loss without detection of a mutation by sequencing is not considered diagnostic of this entity); at a minimum, coverage of exons 4 to 11 (the regions in which the majority of variants are detected) has been recommended.<ref name=":2" /> If a second variant is not detected by sequencing analysis (in which case diagnostic criteria are considered fulfilled, assuming the mutations are not known to affect the same allele) then a technique to assess for copy number loss and/or copy neutral LOH at the ''TP53'' locus is generally required. Fluorescence in situ hybridization with a probe set designed to detect deletions of this locus is a common approach to copy loss detection; alternatives include (but are not limited to) chromosome microarray analysis or next generation sequencing (both of which may also allow for detection of copy neutral LOH) and optical genome mapping. If a qualifying copy number loss or copy neutral LOH cannot be demonstrated, a variant allele fraction above 49% can be considered presumptive evidence of one of these phenomena for diagnostic purposes, presuming a germline ''TP53'' variant is excluded.<ref name=":2" /> | ||
| + | [[File:OGM TP53 loss.tif|thumb|1300x1300px|Identification of copy number loss involving the ''TP53'' locus by optical genome mapping. Left: Detailed copy number track demonstrating loss of 17p (black arrow), including the ''TP53'' locus (red arrow). Right: Circos plot showing the same copy number loss of 17p (black arrow), including the ''TP53'' locus (red arrow), in the context of a complex unbalanced rearrangement involving chromosomes 8, 17, and 20. | ||
| − | + | Image from: Eric McGinnis, Vancouver General Hospital|alt=|none]] | |
| − | |||
==Familial Forms== | ==Familial Forms== | ||
| + | Individuals with germline ''TP53'' variants (as in Li-Fraumeni syndrome) are predisposed to a range of malignancies, including hematologic malignancies. Myeloid malignancies in this population have been reported to commonly occur after cytotoxic therapy for an alternative cancer and to, relatively frequently, also carry a somatic ''TP53'' mutation resulting in biallelic inactivation.<ref name=":2" /><ref>{{Cite journal|last=Swaminathan|first=Mahesh|last2=Bannon|first2=Sarah A.|last3=Routbort|first3=Mark|last4=Naqvi|first4=Kiran|last5=Kadia|first5=Tapan M.|last6=Takahashi|first6=Koichi|last7=Alvarado|first7=Yesid|last8=Ravandi-Kashani|first8=Farhad|last9=Patel|first9=Keyur P.|date=2019-02|title=Hematologic malignancies and Li-Fraumeni syndrome|url=https://pubmed.ncbi.nlm.nih.gov/30709875|journal=Cold Spring Harbor Molecular Case Studies|volume=5|issue=1|pages=a003210|doi=10.1101/mcs.a003210|issn=2373-2873|pmc=6371746|pmid=30709875}}</ref><ref>{{Cite journal|last=Gonzalez|first=Kelly D.|last2=Noltner|first2=Katie A.|last3=Buzin|first3=Carolyn H.|last4=Gu|first4=Dongqing|last5=Wen-Fong|first5=Cindy Y.|last6=Nguyen|first6=Vu Q.|last7=Han|first7=Jennifer H.|last8=Lowstuter|first8=Katrina|last9=Longmate|first9=Jeffrey|date=2009-03-10|title=Beyond Li Fraumeni Syndrome: clinical characteristics of families with p53 germline mutations|url=https://pubmed.ncbi.nlm.nih.gov/19204208|journal=Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology|volume=27|issue=8|pages=1250–1256|doi=10.1200/JCO.2008.16.6959|issn=1527-7755|pmid=19204208}}</ref> | ||
| − | + | Should a myeloid neoplasm meeting diagnostic criteria for MDS-bi''TP53'' develop in an individual with germline predisposition to development of myeloid neoplasms, the appropriate germline predisposition disorder should be added as a diagnostic qualifier: e.g., MDS-bi''TP53'' associated with germline ''TP53'' variant.<ref name=":2" /> | |
| − | |||
==Additional Information== | ==Additional Information== | ||
| + | In the proposed International Consensus Classification system (in which myeloid neoplasms with mutated ''TP53'' were separated as a distinct category), entities meeting diagnostic criteria for MDS-bi''TP53'' using WHO 5th Edition classification standards will generally be compatible with either: myelodysplastic syndrome with mutated ''TP53'', or; myelodysplastic syndrome/acute myeloid leukemia (MDS/AML) with mutated ''TP53''. Key distinctions from WHO 5th Edition diagnostic criteria include:<ref>{{Cite journal|last=Arber|first=Daniel A.|last2=Orazi|first2=Attilio|last3=Hasserjian|first3=Robert P.|last4=Borowitz|first4=Michael J.|last5=Calvo|first5=Katherine R.|last6=Kvasnicka|first6=Hans-Michael|last7=Wang|first7=Sa A.|last8=Bagg|first8=Adam|last9=Barbui|first9=Tiziano|date=2022-09-15|title=International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data|url=https://pubmed.ncbi.nlm.nih.gov/35767897|journal=Blood|volume=140|issue=11|pages=1200–1228|doi=10.1182/blood.2022015850|issn=1528-0020|pmc=9479031|pmid=35767897}}</ref><ref>{{Cite journal|last=Hasserjian|first=Robert P.|last2=Orazi|first2=Attilio|last3=Orfao|first3=Alberto|last4=Rozman|first4=Maria|last5=Wang|first5=Sa A.|date=2023-01|title=The International Consensus Classification of myelodysplastic syndromes and related entities|url=https://pubmed.ncbi.nlm.nih.gov/36287260|journal=Virchows Archiv: An International Journal of Pathology|volume=482|issue=1|pages=39–51|doi=10.1007/s00428-022-03417-1|issn=1432-2307|pmid=36287260}}</ref><ref>{{Cite journal|last=Weinberg|first=Olga K.|last2=Porwit|first2=Anna|last3=Orazi|first3=Attilio|last4=Hasserjian|first4=Robert P.|last5=Foucar|first5=Kathryn|last6=Duncavage|first6=Eric J.|last7=Arber|first7=Daniel A.|date=2023-01|title=The International Consensus Classification of acute myeloid leukemia|url=https://pubmed.ncbi.nlm.nih.gov/36264379|journal=Virchows Archiv: An International Journal of Pathology|volume=482|issue=1|pages=27–37|doi=10.1007/s00428-022-03430-4|issn=1432-2307|pmid=36264379}}</ref> | ||
| − | + | *A VAF threshold of ≥10% is required for each ''TP53'' variant applied in diagnostic criteria | |
| + | *A complex karyotype (at least three distinct abnormalities excluding loss of Y) and/or 17p deletion detected by analysis of banded chromosomes, in the context of one detected ''TP53'' variant, can be considered evidence of biallelic inactivation | ||
| + | *If myeloid blasts account for 10-19% of cellularity in blood or bone marrow then a diagnosis of MDS/AML with mutated ''TP53'' is applied (for which only a single ''TP53'' mutation with VAF ≥10% is required for diagnosis) | ||
==Links== | ==Links== | ||
| − | + | Put your text placeholder here (use "Link" icon at top of page) | |
| − | Put your text placeholder here ( | ||
| − | |||
==References== | ==References== | ||
| − | + | <references /> | |
| − | |||
| − | |||
| − | |||
| − | |||
==Notes== | ==Notes== | ||
Latest revision as of 17:16, 6 September 2024
Haematolymphoid Tumours (WHO Classification, 5th ed.)
Primary Author(s)*
Eric McGinnis, MD, Vancouver General Hospital
WHO Classification of Disease
| Structure | Disease |
|---|---|
| Book | Haematolymphoid Tumours (5th ed.) |
| Category | Myeloid proliferations and neoplasms |
| Family | Myelodysplastic neoplasms |
| Type | Myelodysplastic neoplasms, with defining genetic abnormalities |
| Subtype(s) | Myelodysplastic neoplasm with biallelic TP53 inactivation |
Definition / Description of Disease
Myelodysplastic neoplasm with biallelic TP53 inactivation (MDS-biTP53) is a myeloid neoplasm which is characterized by ineffective hematopoiesis, manifesting clinically and pathologically as cytopenia(s), bone marrow morphologic dysplasia, and propensity to progress to acute myeloid leukemia, and which carries either two TP53 mutations or a single mutation accompanied by either evidence of concurrent TP53 deletion or copy-neutral loss of heterozygosity (LOH); in the appropriate setting, a pathogenic TP53 variant with high variant allele fraction (VAF > 49%) can be considered presumptive evidence of biallelic inactivation status.[1][2]
MDS bi-TP53 supersedes other genetically and morphologically defined MDS entities. By definition, in addition to the requirement for evidence of biallelic TP53 inactivation, general criteria for diagnosis of MDS must be met and diagnostic criteria for acute myeloid leukemia must not be met. Acute erythroid leukemia, in which TP53 mutations are common, must be specifically excluded by morphologic and/or immunophenotypic assessment.[2]
Synonyms / Terminology
Myelodysplastic neoplasm with multi-hit TP53 inactivation, MDS-biTP53
Epidemiology / Prevalence
Inactivating mutations or losses of TP53, whether monoallelic or biallelic, have been reported to occur in approximately 11% of MDS, with a higher frequency in therapy-related MDS (18%) relative to de novo MDS (6%).[3] These genomic aberrations resulting in loss of TP53 are present in a biallelic or multi-hit state in two-thirds of patients with MDS (with a higher reported frequency of 84% in patients with therapy-related disease).[3] The incidence of MDS-biTP53 has been estimated to be approximately 0.03 persons per 100,000 per year.[2]
Clinical Features
Signs and symptoms are typically nonspecific and related to cytopenias (unified thresholds for cytopenias in WHO5 diagnostic categories are detailed below, though these must be interpreted in the context of laboratory reference ranges, patient sex, and relevant comorbidities). TP53 mutations show a strong association with thrombocytopenia in MDS.[3][4]
- Anemia (hemoglobin < 130 g/L in males, < 120 g/L in females)
- Pallor, fatigue
- Neutropenia (absolute neutrophil count < 1.8x109/L)
- Infection
- Thrombocytopenia (platelet count < 150x109/L)
- Bruising, petechiae, bleeding
MDS-biTP53 is a high-risk disease, with increased risk of leukemic transformation and decreased overall survival; this elevated risk is independent of IPSS-R risk stratification and persists in the context of allogeneic hematopoietic stem cell transplantation. In the absence of comprehensive assessment for copy number losses or copy neutral LOH affecting the TP53 locus, evidence suggests that the presence of a TP53 mutation at high (>40%) VAF, particularly in the context of a complex karyotype, may be associated with a similarly poor prognosis to MDS-biTP53.[3][2][5][6]
| Signs and Symptoms | Pallor
Fatigue Infection Bruising Bleeding Petechiae |
| Laboratory Findings | Anemia (hemoglobin < 130 g/L in males, < 120 g/L in females)
Neutropenia (neutrophils < 1.8x109/L) Thrombocytopenia (platelets < 150x109/L) Circulating blasts (+/-) Elevated lactate dehydrogenase (+/-) |
Sites of Involvement
Bone marrow and peripheral blood; rarely, extramedullary proliferations occur.[7]
Morphologic Features
Morphologic dysplasia is the hallmark of MDS, including MDS-biTP53; by definition, at least one lineage . TP53 mutations in MDS have been associated with particularly severe granulocytic dysplasia and with increased frequency of bone marrow blasts.[3][4][8] TP53 mutations are enriched in MDS associated with bone marrow fibrosis.[9][10]
| Hematopoietic lineage | Characteristic dysplastic features |
|---|---|
| Erythroid | Nuclear budding
Internuclear bridging Multinuclearity Karyorrhexis Megaloblastosis Cytoplasmic vacuolation Ring sideroblasts Periodic acid Schiff positivity |
| Granulocytic | Nuclear hyposegmentation (Pelgeroid changes)
Nuclear hypersegmentation Cytoplasmic hypogranularity Abnormally small cell size Auer rods Pseudo-Chédiak–Higashi granules |
| Megakaryocytic | Nuclear hypolobation
Widely separated nuclear lobes Micromegakaryocytes |
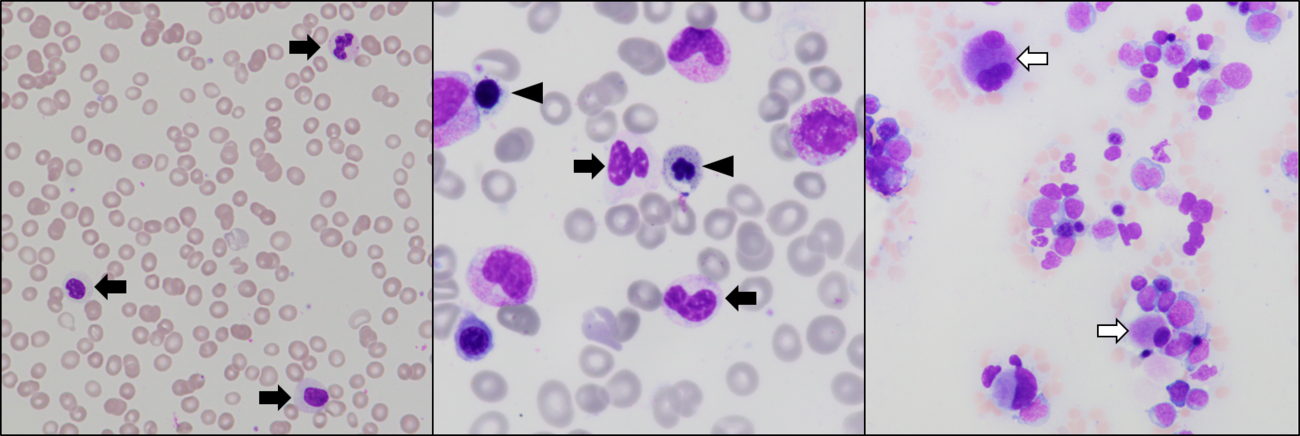
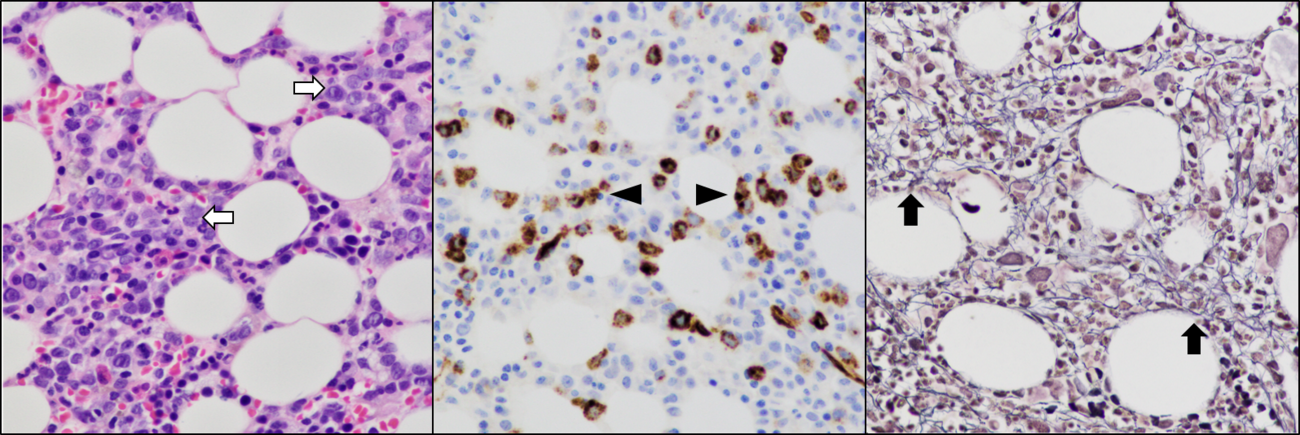
Immunophenotype
Immunophenotyping by flow cytometry using assays designed to detect abnormal numbers, light scatter characteristics, and antigen expression patterns of hematopoietic cells can provide useful data supportive of a diagnosis of MDS (and aid in blast enumeration); however, these findings are not specific to MDS-biTP53 and are not formalized in current diagnostic criteria.[2][11]
Immunohistochemical evaluation of TP53 protein expression in bone marrow specimens can be used to inform TP53 mutation status. Several studies evaluating TP53 protein expression in bone marrow specimens have consistently demonstrated nuclear overexpression (thought to result from stabilization of TP53 resulting from dominant negative mutations), when present in an increased fraction of cells, to be associated with pathogenic TP53 mutation and to be similarly predictive of poor outcomes.[12][13][14][15][16] Complete absence of TP53 expression demonstrable by immunohistochemistry has similarly been associated with TP53 mutation (typically frameshift, nonsense, and splice site, often with coexisting copy loss).[12]
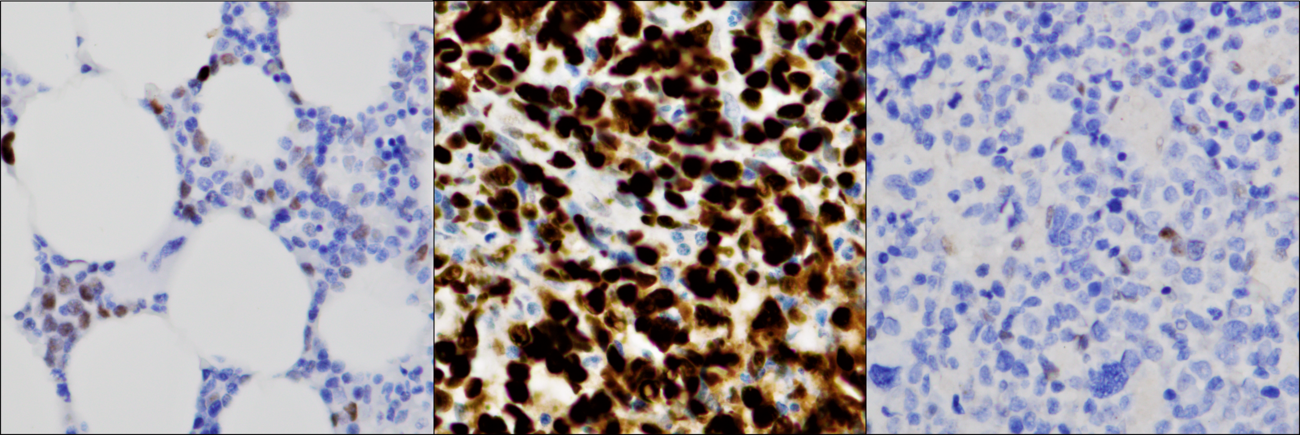
| Positive (universal) | N/A |
| Positive (subset) | N/A |
| Negative (universal) | N/A |
| Negative (subset) | N/A |
Chromosomal Rearrangements (Gene Fusions)
There are no known recurrent rearrangements resulting in gene fusions associated with MDS-biTP53.
| Chromosomal Rearrangement | Genes in Fusion
(5’ or 3’ Segments) |
Pathogenic Derivative | Prevalence | Diagnostic Significance (Yes, No or Unknown) | Prognostic Significance (Yes, No or Unknown) | Therapeutic Significance (Yes, No or Unknown) | Notes |
| EXAMPLE: t(9;22)(q34;q11.2) | EXAMPLE: 3'ABL1 / 5'BCR | EXAMPLE: der(22) | EXAMPLE: 20% (COSMIC)
EXAMPLE: 30% (add reference) |
Yes | No | Yes | EXAMPLE:
The t(9;22) is diagnostic of CML in the appropriate morphology and clinical context (add reference). This fusion is responsive to targeted therapy such as Imatinib (Gleevec) (add reference). |
Individual Region Genomic Gain/Loss/LOH
MDS bi-TP53 is strongly associated with karyotype complexity, with a higher average number of cytogenetic abnormalities (rearrangements, copy gains, and copy losses) relative to MDS with unmutated TP53 or with monoallelic mutation, with particular predilection for recurrent copy number losses.[4][3] Recurrent chromosomal gains, losses, and regions of LOH described which have been observed at higher frequency in MDS-biTP53 include[3][2][4]:
Loss / Deletion
- Deletion 5q (most common co-occurring abnormality)
- Loss of chromosome 7 or deletion 7q (second most common co-occurring abnormality)
- Deletion 17p
- Deletion 12p
- Losses involving chromosome 13
- Losses involving chromosome 18
- Deletion 20q
Gain
- Trisomy 8
- Trisomy 21
- Gain of 1p
- Trisomy 22
- Gain of 11q
Copy-neutral LOH involving chromosome 17p and the TP53 locus is a component of the diagnostic definition of MDS-biTP53 and has been reported to occur in 21% of patients with this disease, usually in the setting of a single TP53 mutation (i.e. not typically in association with more than one distinct TP53 mutation).[3] It is not currently known whether other instances of recurrent copy-neutral LOH in myeloid neoplasms are observed at significantly higher frequency in MDS-biTP53.
| Chr # | Gain/Loss/Amp/LOH | Minimal Region Genomic Coordinates [Genome Build] | Minimal Region Cytoband | Diagnostic Significance (Yes, No or Unknown) | Prognostic Significance
(Yes, No or Unknown) |
Therapeutic Significance
(Yes, No or Unknown) |
Notes |
| 1 | Gain | 1p | No | No* | No | ||
| 5 | Loss | 5q | No | No* | No | Most common co-occurring abnormality | |
| 7 | Loss | chr7 / 7q | No | No* | No | Second most common co-occurring abnormality | |
| 8 | Gain | chr8 | No | No* | No | ||
| 11 | Gain | 11q | No | No* | No | ||
| 12 | Loss | 12p | No | No* | No | ||
| 13 | Loss | 13q | No | No* | No | ||
| 17 | Loss / LOH | 17p | Yes | Yes** | Yes | Key component of entity classification | |
| 18 | Loss | No | No* | No | |||
| 20 | Loss | 20q | No | No* | No | ||
| 21 | Gain | chr21 | No | No* | No | ||
| 22 | Gain | chr22 | No | No* | No |
*Adverse prognosis associated with MDS-biTP53 appears to persist independent of IPSS-R status
**MDS-biTP53 diagnosis and therefore prognostic stratification is dependent on loss or LOH involving 17p unless multiple mutations are present
Characteristic Chromosomal Patterns
MDS-biTP53 is strongly associated with complex (and monosomal) karyotypes; patients with biallelic inactivation of TP53 showed significantly higher numbers of distinct cytogenetic abnormalities (median six excluding chromosome 17) relative to patients with monoallelic inactivation (median one), and a large majority of these tend to be accounted for by abnormalities associated with genomic loss (deletion/monosomy), with a median of four such abnormalities in MDS-biTP53, which are particularly enriched for deletions involving chromosome 5q (identified in 85% of patients).[3][2][4] Abnormalities involving chromosome 17p resulting in TP53 loss are, by virtue of diagnostic criteria, present in approximately one third of MDS bi-TP53, in which they may result from simple deletion or unbalanced rearrangement, and which may in cases be submicroscopic/cryptic to analysis of banded chromosomes.[3]
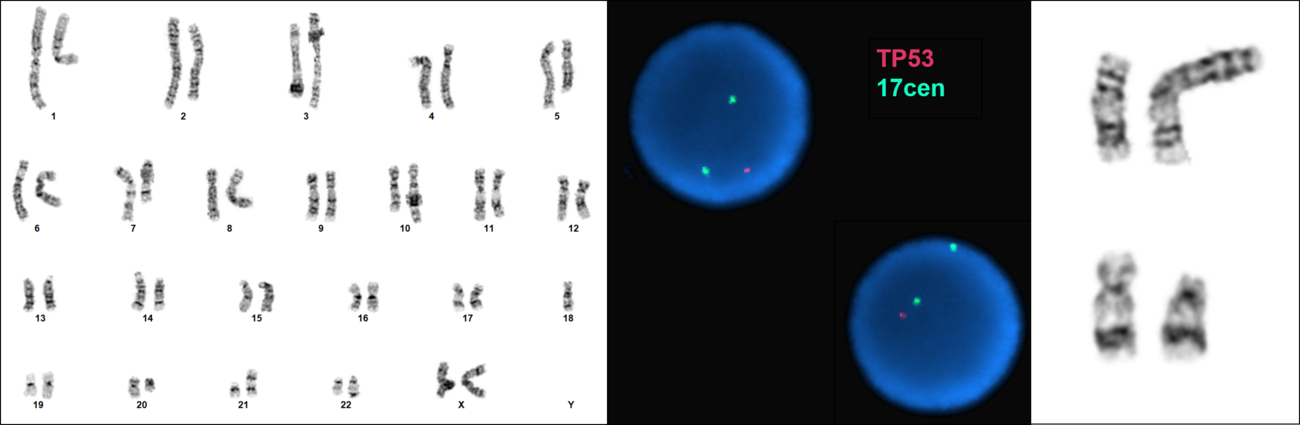
| Chromosomal Pattern | Diagnostic Significance (Yes, No or Unknown) | Prognostic Significance
(Yes, No or Unknown) |
Therapeutic Significance
(Yes, No or Unknown) |
Notes |
| Complex karyotype | No (see note) | Yes (see note) | No (see note) | Often with abnormalities resulting in chromosomal losses, especially involving chromosome 5q and chromosome 7
Complex karyotypes are strongly associated with MDS-biTP53 and, in the absence of available data for identification of LOH at the TP53 locus, may be suggestive of biallelic inactivation and may be associated with similarly poor prognosis |
Gene Mutations (SNV/INDEL)
At least one mutation involving TP53 is required to establish a diagnosis of MDS biTP53; a second distinct mutation involving TP53, fulfilling diagnostic criteria without deletion or copy neutral LOH, is identified in approximately one third of patients with MDS-biTP53.[2][3] TP53 mutations reported in MDS-biTP53 show a similar pattern of distribution to those previously reported, primarily clustering in the DNA-binding domain with smaller numbers of (primarily truncating) mutations upstream (extending to the transactivation domain) or downstream (extending downstream of the oligomerization domain); available data suggest approximately 70% of mutations to be missense variants, 28% (a higher frequency than was observed in MDS with monoallelic TP53 mutation) to be truncating (including nonsense, frameshift, splice site, and nonstop variants), and a small fraction to be in-frame deletions or insertions.[3] In MDS-biTP53, 20% of TP53 variants identified were accounted for by hotspot mutations affecting amino acids 273, 220, 248, and 175 (in order of descending frequency). Variants identified in the context of multiple distinct TP53 mutations have been reported with a median VAF of 32% (range: 2% to 52%) while those associated with a second hit resulting from TP53 locus deletion or copy-neutral LOH have a reported median VAF of 41% (2% to 92% with a bimodal distribution) and 71% (2% to 98%), respectively.[3]
TP53-mutated MDS, in general, tends to carry fewer cooperating somatic mutations that MDS without TP53 mutation; MDS bi-TP53 is further associated with fewer cooperating somatic mutations than MDS with monoallelic inactivation of TP53, with 40% of MDS-biTP53 reported to show no additional identifiable driver mutations.[3][4] Genes recurrently mutated in this group are similar to those described in MDS in general, and include DNMT3A, TET2, ASXL1, U2AF1, PPM1D, EZH2, SF3B1, NF1; infrequently, comutations identified include BCOR, JAK2, and CBL. MDS carrying at least one mutation in TP53, as a whole, has been observed to less frequently carry mutations in ASXL1, U2AF1, and RUNX1, while MDS-biTP53 has been observed to have lower rates of TET2, SF3B1, ASXL1, SRSF2, and RUNX1 mutation relative to MDS with a single TP53 mutation.[3][4]
Note: A more extensive list of mutations can be found in cBioportal (https://www.cbioportal.org/), COSMIC (https://cancer.sanger.ac.uk/cosmic), ICGC (https://dcc.icgc.org/) and/or other databases. When applicable, gene-specific pages within the CCGA site directly link to pertinent external content.
| Gene; Genetic Alteration | Presumed Mechanism (Tumor Suppressor Gene (TSG)/Oncogene/Other) | Prevalence (COSMIC/ TCGA/Other) | Concomitant Mutations | Mutually Exclusive Mutations | Diagnostic Significance (Yes, No or Unknown) | Prognostic Significance
(Yes, No or Unknown) |
Therapeutic Significance
(Yes, No or Unknown) |
Notes |
| TP53; LOF and/or GOF mutations | Tumor Suppressor Gene / Oncogene (GOF variants) | 100% (entity-defining for MDS-biTP53) | DNMT3A, TET2, ASXL1, U2AF1, PPM1D, EZH2, SF3B1, NF1 (see note) | None | Yes | Yes | Yes (see note) | MDS-biTP53 carries, on average, less cooperating somatic mutations than MDS without TP53 mutation
MDS-biTP53 is associated with poorer responses to most therapeutic modalities |
Epigenomic Alterations
Similar to other MDS entities, mutations in chromatin-modifying/epigenetic regulator genes (e.g. TET2, EZH2, DNMT3A, ASXL1) are recurrent in MDS-biTP53, albeit at a lower frequency. Certain TP53 mutations, including hotspot mutations identified in MDS-biTP53, have been demonstrated to confer gain-of-function effects to the mutant TP53, some of which appear to be mediated through modification of the activities of chromatin regulatory/epigenetic modifier genes such as KMT2A, KMT2D, and EZH2.[17][18]
Genes and Main Pathways Involved
p53, a tumour repressor transcription factor encoded by the TP53 gene, is central to regulation of a complex network of pathways by which cells respond to intra- or extracellular stressors, including cell cycle progression and response to hyperproliferation signals, cell survival and apoptosis, and maintenance of genome stability.[19][20][21] Mutations inactivating TP53 typically result in disruption of normal tumour suppressor activities of the protein and, depending on the cell state and mutational profile, may exert dominant negative effects on residual wild-type p53 and/or may confer neomorphic oncogenic function to the protein.[22][23] p53 is required for genomic stability and mutations affecting TP53 are among the most frequent abnormalities identified in human malignancies, in which they are strongly associated with genomic instability (in the form of increased frequency of aneuploidy, polyploidy/tetraploidy, and chromosomal rearrangements).[3][24]
| Gene; Genetic Alteration | Pathway | Pathophysiologic Outcome |
| TP53; Inactivating +/- Gain of abnormal function mutations | TP53 signaling network; cell cycle arrest, DNA repair, cell senescence, apoptosis in response to DNA damage, oncogene activation, cell stressors | Failures of DNA repair, cell cycle arrest, apoptosis in response to DNA damage, hyperproliferation, or cell stressors |
Genetic Diagnostic Testing Methods
As classification of MDS-biTP53 requires demonstration of at least one TP53 mutation, sequencing analysis is required in all cases (i.e. cytogenetic evidence supportive of TP53 loss without detection of a mutation by sequencing is not considered diagnostic of this entity); at a minimum, coverage of exons 4 to 11 (the regions in which the majority of variants are detected) has been recommended.[2] If a second variant is not detected by sequencing analysis (in which case diagnostic criteria are considered fulfilled, assuming the mutations are not known to affect the same allele) then a technique to assess for copy number loss and/or copy neutral LOH at the TP53 locus is generally required. Fluorescence in situ hybridization with a probe set designed to detect deletions of this locus is a common approach to copy loss detection; alternatives include (but are not limited to) chromosome microarray analysis or next generation sequencing (both of which may also allow for detection of copy neutral LOH) and optical genome mapping. If a qualifying copy number loss or copy neutral LOH cannot be demonstrated, a variant allele fraction above 49% can be considered presumptive evidence of one of these phenomena for diagnostic purposes, presuming a germline TP53 variant is excluded.[2]
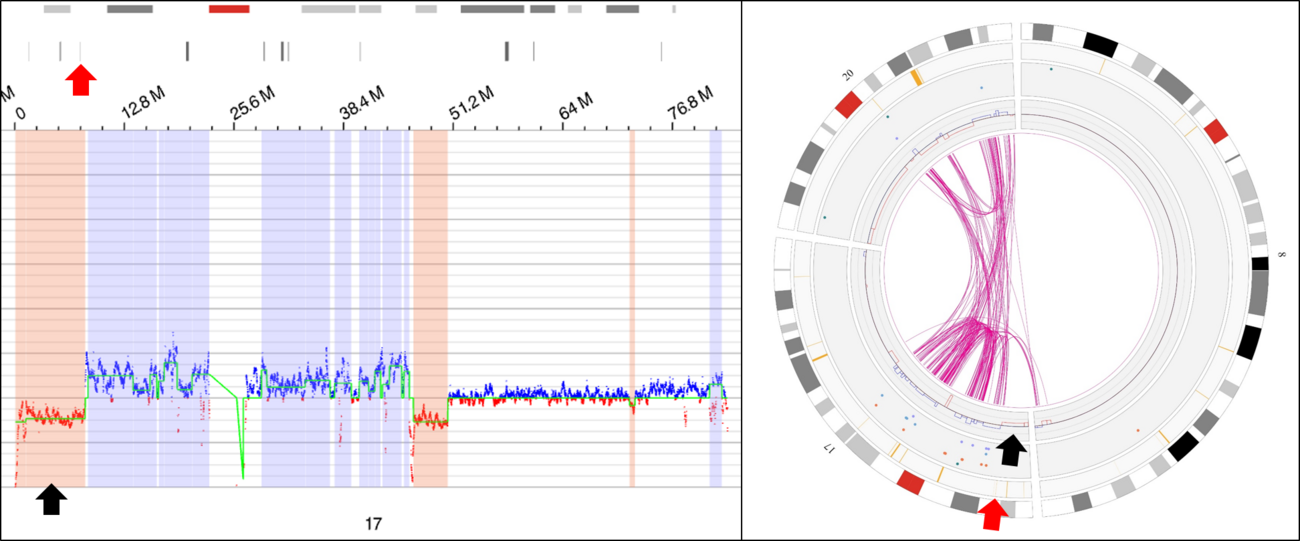
Familial Forms
Individuals with germline TP53 variants (as in Li-Fraumeni syndrome) are predisposed to a range of malignancies, including hematologic malignancies. Myeloid malignancies in this population have been reported to commonly occur after cytotoxic therapy for an alternative cancer and to, relatively frequently, also carry a somatic TP53 mutation resulting in biallelic inactivation.[2][25][26]
Should a myeloid neoplasm meeting diagnostic criteria for MDS-biTP53 develop in an individual with germline predisposition to development of myeloid neoplasms, the appropriate germline predisposition disorder should be added as a diagnostic qualifier: e.g., MDS-biTP53 associated with germline TP53 variant.[2]
Additional Information
In the proposed International Consensus Classification system (in which myeloid neoplasms with mutated TP53 were separated as a distinct category), entities meeting diagnostic criteria for MDS-biTP53 using WHO 5th Edition classification standards will generally be compatible with either: myelodysplastic syndrome with mutated TP53, or; myelodysplastic syndrome/acute myeloid leukemia (MDS/AML) with mutated TP53. Key distinctions from WHO 5th Edition diagnostic criteria include:[27][28][29]
- A VAF threshold of ≥10% is required for each TP53 variant applied in diagnostic criteria
- A complex karyotype (at least three distinct abnormalities excluding loss of Y) and/or 17p deletion detected by analysis of banded chromosomes, in the context of one detected TP53 variant, can be considered evidence of biallelic inactivation
- If myeloid blasts account for 10-19% of cellularity in blood or bone marrow then a diagnosis of MDS/AML with mutated TP53 is applied (for which only a single TP53 mutation with VAF ≥10% is required for diagnosis)
Links
Put your text placeholder here (use "Link" icon at top of page)
References
- ↑ Khoury, Joseph D.; et al. (2022-07). "The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms". Leukemia. 36 (7): 1703–1719. doi:10.1038/s41375-022-01613-1. ISSN 1476-5551. PMC 9252913 Check
|pmc=value (help). PMID 35732831 Check|pmid=value (help). Check date values in:|date=(help) - ↑ 2.00 2.01 2.02 2.03 2.04 2.05 2.06 2.07 2.08 2.09 2.10 2.11 Germing U., Claudi M., Zerbini N., et al. Myelodysplastic neoplasm with biallelic TP53 inactivation. In: WHO Classification of Tumours Editorial Board. Haematolymphoid tumours [Internet; beta version ahead of print]. Lyon (France): International Agency for Research on Cancer; 2022 [cited YYYY Mmm D]. (WHO classification of tumours series, 5th ed.; vol. 11). Available from: https://tumourclassification.iarc.who.int/chapters/63.
- ↑ 3.00 3.01 3.02 3.03 3.04 3.05 3.06 3.07 3.08 3.09 3.10 3.11 3.12 3.13 3.14 3.15 Bernard, Elsa; et al. (2020-10). "Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes". Nature Medicine. 26 (10): 1549–1556. doi:10.1038/s41591-020-1008-z. ISSN 1546-170X. PMC 8381722 Check
|pmc=value (help). PMID 32747829 Check|pmid=value (help). Check date values in:|date=(help) - ↑ 4.0 4.1 4.2 4.3 4.4 4.5 4.6 Haase, Detlef; et al. (2019-07). "TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups". Leukemia. 33 (7): 1747–1758. doi:10.1038/s41375-018-0351-2. ISSN 1476-5551. PMC 6609480. PMID 30635634. Check date values in:
|date=(help) - ↑ Sallman, D. A.; et al. (2016-03). "Impact of TP53 mutation variant allele frequency on phenotype and outcomes in myelodysplastic syndromes". Leukemia. 30 (3): 666–673. doi:10.1038/leu.2015.304. ISSN 1476-5551. PMC 7864381 Check
|pmc=value (help). PMID 26514544. Check date values in:|date=(help) - ↑ Grob, Tim; et al. (2022-04-14). "Molecular characterization of mutant TP53 acute myeloid leukemia and high-risk myelodysplastic syndrome". Blood. 139 (15): 2347–2354. doi:10.1182/blood.2021014472. ISSN 1528-0020. PMID 35108372 Check
|pmid=value (help). - ↑ Fan, N.; et al. (2018-11-19). "Extramedullary hematopoiesis in the absence of myeloproliferative neoplasm: Mayo Clinic case series of 309 patients". Blood Cancer Journal. 8 (12): 119. doi:10.1038/s41408-018-0156-6. ISSN 2044-5385. PMC 6242913. PMID 30455416.
- ↑ Della Porta, M. G.; et al. (2015-01). "Minimal morphological criteria for defining bone marrow dysplasia: a basis for clinical implementation of WHO classification of myelodysplastic syndromes". Leukemia. 29 (1): 66–75. doi:10.1038/leu.2014.161. ISSN 1476-5551. PMID 24935723. Check date values in:
|date=(help) - ↑ Duarte, Fernando B.; et al. (2018-05). "Bone marrow fibrosis at diagnosis is associated with TP53 overexpression and adverse prognosis in low-risk myelodysplastic syndrome". British Journal of Haematology. 181 (4): 547–549. doi:10.1111/bjh.14656. ISSN 1365-2141. PMID 28318026. Check date values in:
|date=(help) - ↑ Loghavi, Sanam; et al. (2015-10). "TP53 overexpression is an independent adverse prognostic factor in de novo myelodysplastic syndromes with fibrosis". British Journal of Haematology. 171 (1): 91–99. doi:10.1111/bjh.13529. ISSN 1365-2141. PMC 5577911. PMID 26123119. Check date values in:
|date=(help) - ↑ Kern, Wolfgang; et al. (2023-01). "Multicenter prospective evaluation of diagnostic potential of flow cytometric aberrancies in myelodysplastic syndromes by the ELN iMDS flow working group". Cytometry. Part B, Clinical Cytometry. 104 (1): 51–65. doi:10.1002/cyto.b.22105. ISSN 1552-4957. PMID 36416672 Check
|pmid=value (help). Check date values in:|date=(help) - ↑ 12.0 12.1 Tashakori, Mehrnoosh; et al. (2022-07-07). "TP53 copy number and protein expression inform mutation status across risk categories in acute myeloid leukemia". Blood. 140 (1): 58–72. doi:10.1182/blood.2021013983. ISSN 1528-0020. PMC 9346958 Check
|pmc=value (help). PMID 35390143 Check|pmid=value (help). - ↑ Saft, Leonie; et al. (2014-06). "p53 protein expression independently predicts outcome in patients with lower-risk myelodysplastic syndromes with del(5q)". Haematologica. 99 (6): 1041–1049. doi:10.3324/haematol.2013.098103. ISSN 1592-8721. PMC 4040908. PMID 24682512. Check date values in:
|date=(help) - ↑ Loghavi, Sanam; et al. (2015-10). "TP53 overexpression is an independent adverse prognostic factor in de novo myelodysplastic syndromes with fibrosis". British Journal of Haematology. 171 (1): 91–99. doi:10.1111/bjh.13529. ISSN 1365-2141. PMC 5577911. PMID 26123119. Check date values in:
|date=(help) - ↑ Ruzinova, Marianna B.; et al. (2019-08). "TP53 immunohistochemistry correlates with TP53 mutation status and clearance in decitabine-treated patients with myeloid malignancies". Haematologica. 104 (8): e345–e348. doi:10.3324/haematol.2018.205302. ISSN 1592-8721. PMC 6669137. PMID 30792212. Check date values in:
|date=(help) - ↑ Kubbutat, M. H.; et al. (1997-05-15). "Regulation of p53 stability by Mdm2". Nature. 387 (6630): 299–303. doi:10.1038/387299a0. ISSN 0028-0836. PMID 9153396.
- ↑ Zhu, Jiajun; et al. (2015-09-10). "Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth". Nature. 525 (7568): 206–211. doi:10.1038/nature15251. ISSN 1476-4687. PMC 4568559. PMID 26331536.
- ↑ Chen, Sisi; et al. (2019-12-11). "Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway". Nature Communications. 10 (1): 5649. doi:10.1038/s41467-019-13542-2. ISSN 2041-1723. PMC 6906427. PMID 31827082.
- ↑ Eischen, Christine M. (2016-06-01). "Genome Stability Requires p53". Cold Spring Harbor Perspectives in Medicine. 6 (6): a026096. doi:10.1101/cshperspect.a026096. ISSN 2157-1422. PMC 4888814. PMID 27252396.
- ↑ Levine, Arnold J.; et al. (2009-10). "The first 30 years of p53: growing ever more complex". Nature Reviews. Cancer. 9 (10): 749–758. doi:10.1038/nrc2723. ISSN 1474-1768. PMC 2771725. PMID 19776744. Check date values in:
|date=(help) - ↑ Boettcher, Steffen; et al. (2019-08-09). "A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies". Science (New York, N.Y.). 365 (6453): 599–604. doi:10.1126/science.aax3649. ISSN 1095-9203. PMC 7327437 Check
|pmc=value (help). PMID 31395785. - ↑ Stein, Yan; et al. (2020-12-31). "Mutant p53 oncogenicity: dominant-negative or gain-of-function?". Carcinogenesis. 41 (12): 1635–1647. doi:10.1093/carcin/bgaa117. ISSN 1460-2180. PMID 33159515 Check
|pmid=value (help). - ↑ Boettcher, Steffen; et al. (2019-08-09). "A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies". Science (New York, N.Y.). 365 (6453): 599–604. doi:10.1126/science.aax3649. ISSN 1095-9203. PMC 7327437 Check
|pmc=value (help). PMID 31395785. - ↑ Eischen, Christine M. (2016-06-01). "Genome Stability Requires p53". Cold Spring Harbor Perspectives in Medicine. 6 (6): a026096. doi:10.1101/cshperspect.a026096. ISSN 2157-1422. PMC 4888814. PMID 27252396.
- ↑ Swaminathan, Mahesh; et al. (2019-02). "Hematologic malignancies and Li-Fraumeni syndrome". Cold Spring Harbor Molecular Case Studies. 5 (1): a003210. doi:10.1101/mcs.a003210. ISSN 2373-2873. PMC 6371746. PMID 30709875. Check date values in:
|date=(help) - ↑ Gonzalez, Kelly D.; et al. (2009-03-10). "Beyond Li Fraumeni Syndrome: clinical characteristics of families with p53 germline mutations". Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology. 27 (8): 1250–1256. doi:10.1200/JCO.2008.16.6959. ISSN 1527-7755. PMID 19204208.
- ↑ Arber, Daniel A.; et al. (2022-09-15). "International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data". Blood. 140 (11): 1200–1228. doi:10.1182/blood.2022015850. ISSN 1528-0020. PMC 9479031 Check
|pmc=value (help). PMID 35767897 Check|pmid=value (help). - ↑ Hasserjian, Robert P.; et al. (2023-01). "The International Consensus Classification of myelodysplastic syndromes and related entities". Virchows Archiv: An International Journal of Pathology. 482 (1): 39–51. doi:10.1007/s00428-022-03417-1. ISSN 1432-2307. PMID 36287260 Check
|pmid=value (help). Check date values in:|date=(help) - ↑ Weinberg, Olga K.; et al. (2023-01). "The International Consensus Classification of acute myeloid leukemia". Virchows Archiv: An International Journal of Pathology. 482 (1): 27–37. doi:10.1007/s00428-022-03430-4. ISSN 1432-2307. PMID 36264379 Check
|pmid=value (help). Check date values in:|date=(help)
Notes
*Primary authors will typically be those that initially create and complete the content of a page. If a subsequent user modifies the content and feels the effort put forth is of high enough significance to warrant listing in the authorship section, please contact the CCGA coordinators (contact information provided on the homepage). Additional global feedback or concerns are also welcome. *Citation of this Page: “Myelodysplastic neoplasm with biallelic TP53 inactivation”. Compendium of Cancer Genome Aberrations (CCGA), Cancer Genomics Consortium (CGC), updated 09/6/2024, https://ccga.io/index.php/HAEM5:Myelodysplastic_neoplasm_with_biallelic_TP53_inactivation.